A chromosome-level genome assembly of the European green toad (Bufotes viridis)
Patrik Rödin-Mörch, Ignas Bunikis, Eunkyoung Choi, Claudio Ciofi, Genevieve Diedericks, Maria Angela Diroma, Elísabet Einarsdóttir, Kristofer Försäter, Julia Heintz, Linnea Jonsäll, Henrik Lantz, Anssi Laurila, Henrique G Leitão, Mai-Britt Mosbech, Chiara Natali

TL;DR
This paper presents a high-quality chromosome-level genome assembly for the European green toad to aid conservation and genomic research.
Contribution
The novel contribution is the creation of a chromosome-level reference genome for Bufotes viridis.
Findings
The genome assembly includes 11 chromosomes and has a scaffold N50 of 478.39 Mb.
It covers 90.4% of single copy tetrapod orthologs and predicts 23,830 protein-coding genes.
The assembly will support conservation genetics and evolutionary genomics studies in amphibians.
Abstract
The European green toad (Bufotes viridis) is geographically widely distributed. While the species global conservation status is labeled as of least concern by the IUCN, it is declining in many parts of its range where populations are fragmented and isolated. A high-quality reference genome is an important resource for conservation genomic researchers who are trying to understand and interpret the genomic signals of population decline, inbreeding, and the accumulation of deleterious mutations. Here, we assembled and annotated a chromosome-level reference genome for B. viridis as part of the European Reference Genome Atlas pilot project. The genome assembly, with a size of ∼3.89 Gb consists of 11 chromosomes and an additional 2,096 unplaced scaffolds. The final assembly had a scaffold N50 value of 478.39 Mb and covered 90.4% single copy tetrapod orthologs, and 46.7% repetitive elements.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
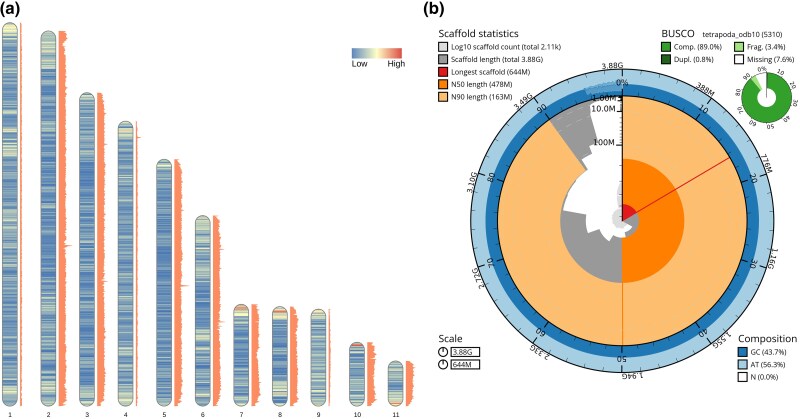 Fig. 1
Fig. 1| Assembly statistics | |
|---|---|
| Contig L50 | 168 |
| Contig N50 (Mb) | 6.73 |
| Longest contig (Mb) | 37 |
| Scaffold L50 | 3 |
| Scaffold N50 (Mb) | 478.39 |
| Longest scaffold (Mb) | 643.6 |
| Number of scaffolds | 2,107 |
| Annotation statistics | |
| Number of protein-coding genes | 31,608 |
| Number of mRNA | 69,426 |
| Number of single exon genes | 7,746 |
| Average number of exons per mRNA | 7.9 |
| Average exon length | 310 |
| Average intron length | 4,984 |
| Repeat statistics | |
| DNA_transposons | 366,392 |
| LINE | 178,322 |
| LTR | 110,821 |
| LOW_complexity | 183,696 |
| RC | 1,348 |
| Retroposon | 41 |
| SINE | 2,398 |
| Satellite | 3,213 |
| Simple_repeat | 1,016,805 |
| Unknown | 2,005,319 |
| rRNA | 1,845 |
| scRNA | 1 |
| snRNA | 18,749 |
| srpRNA | 1 |
| tRNA | 949 |
- —European Reference Genome Atlas (ERGA) consortium10.13039/100020931
- —Biodiversity Genomics Europe project
- —Horizon Europe10.13039/100018693
- —Biodiversity, Circular Economy and Environment call
- —Swiss State Secretariat for Education, Research and Innovation10.13039/501100007352
- —UK Research and Innovation10.13039/100014013
- —Department for Business, Energy and Industrial Strategy10.13039/100011693
- —Uppsala University10.13039/501100007051
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Amphibian and Reptile Biology · Aquaculture disease management and microbiota
Introduction
Amphibians are considered to be the most threatened group of vertebrates, with more than 40% of the assessed amphibian species being threatened with extinction as a result of infectious disease, habitat loss, and climate change (Luedtke et al. 2023). Conservation genomics provides an approach for understanding basic biological properties relevant for the conservation of threatened taxa such as genetic diversity, population structure, amount of inbreeding, and genetic load (Formenti et al. 2022; Theissinger et al. 2023). As such, a high-quality reference genome is a valuable asset facilitating such work.
Among vertebrates, amphibians have the largest variance in genome size, ranging from 140 pg to as small as 0.95 pg (Liedtke et al. 2018). In anurans, the order consisting of frogs and toads, evidence suggests that the wide range in genome sizes is largely due to the recent accumulation of transposable elements, as well as the failure to remove ancient transposable element insertions (Zuo et al. 2023). Due to their genome size and complexity, amphibians have until fairly recently been largely overlooked by large scale initiatives to produce reference genomes, with only a small fraction of the ∼8,600 described amphibian species having a chromosome-level genome assembly available (Kosch et al. 2024).
The green toad genus Bufotes is geographically wide-spread and contains up of 15 recognized lineages. These include lineages that are diploid or allopolyploids (the latter having formed through hybrid speciation and that occurs to the eastern part of the Bufotes range) (Dufresnes et al. 2019). The Bufotes viridis subgroup occurs from Western Europe and the Northern part of the Mediterranean region to the Ural Mountains in the east, and is globally listed as of least concern but decreasing throughout its range (IUCN 2023). For example, in Sweden B. viridis has historically been declining and is considered to be the most vulnerable amphibian species in the country (Höglund et al. 2022), with its entire range confined to fragmented populations along the southern coast or on rocky islands off the coast, which are all geographically isolated from the European mainland populations.
Here, we present a chromosome-level, annotated reference genome for the pilot species B. viridis in the European Reference Genome Atlas (ERGA) pilot project (Mc Cartney et al. 2024). This reference genome will add to the growing number of amphibian reference genomes that will serve as useful tools for comparative and eco-evolutionary genomics studies, and to be used directly or indirectly to aid conservation practitioners working to safeguard amphibian diversity.
Methods
DNA and RNA extractions, sequencing library preparations, and sequencing
One male B. viridis from the ex situ breeding program at Nordens Ark (https://nordensark.se/bevarande/bevarande-i-sverige/gronflackig-padda/) was humanely euthanized and dissected by a veterinarian on the 2021 May 11 (Supplementary Fig. S1) with the approval from the Ethical committee for animal experiments in Göteborg (Dnr 5.8.18-06182/2021). HMW-DNA for PacBio Sequel II sequencing, frozen ground tissue for Omni-C and RNA for Illumina RNA-seq and PacBio Iso-Seq using 7 tissues (skin, spleen, breast muscle, lung, heart, liver, and kidney) were sequenced (See Supplementary Material online for details).
Genome assembly
The PacBio HiFi reads were investigated for potential contamination using Mash Screen v2.3 (Ondov et al. 2019) against the NCBI RefSeq database (https://gembox.cbcb.umd.edu/mash/refseq.genomes.k21s1000.msh), and no contamination was detected. Further evaluation of coverage, genome size, and heterozygosity based on the HiFi reads was done using GeneScopeFK (https://github.com/thegenemyers/GENESCOPE.FK) using a k-mer size of 31, which is a modified version of GenomeScope v2.0 (Ranallo-Benavidez et al. 2020).
The PacBio HiFi https://www.doi.org/10.5281/zenodo.3552717 reads were assembled using IPA v.1.5.0 (https://github.com/PacificBiosciences/pbipa). The initial assembly, the purged assembly and the scaffolded assembly were evaluated with BUSCO v5.4.6 (Manni et al. 2021) and K-mer frequency histogram and completeness statistics produced using MERQURY.FK (https://github.com/thegenemyers/MERQURY.FK), as well as general assembly summary statistics using QUAST v.5.0.2 (Gurevich et al. 2013) (Table 1). The HiFi assembly was purged of duplicates using the IPA internal purge_dups v.1.2.5 (Guan et al. 2020). Omni-C reads were mapped to the contig assembly using BWA v. 0.7.17-r1188 (Li and Durbin 2009), with parameters −5SP and -T0. The alignments were then filtered and processed using pairtools v.0.3.0 (Open2C; Abdennur et al. 2023). The contig assembly was subsequently scaffolded using YaHS v.1.2.a.1.patch (Zhou et al. 2022). Manual curation of the scaffolded assembly was performed in Juicebox v.2.16.00 (Durand et al. 2016), and finalized using the juicer post command in YaHS.
Genome annotation
Using the Nextflow pipeline AnnotationPreprocessing (https://github.com/NBISweden/pipelines-nextflow), an initial annotation preprocessing step was performed by assessing assembly statistics, presence of missing and ambiguous nucleotides, gene completeness, presence of organelles, and distance to other annotated species.
Curated protein sequences were downloaded from the UniProt database (Magrane and UniProt Consortium 2011, downloaded on 2022-12; 568,363 proteins), including the amphibian UniProt database (38,940 proteins, uniprot_amphibia_PE12.fasta). A species specific repeat library was created using RepeatModeler v.2.0.2a (Smit, Hubley and Green 2010; Flynn et al. 2020) and RepeatRunner (Yandell 2006). The identified repeats (excluding transposons), were then matched against the collected protein set in order to exclude nucleotide motifs originating from low-complexity coding sequences. Repetitive sequences were then masked in the genome using the identified repeats with RepeatMasker v.4.1.2_p1 (Smit and Hubley 2010). Illumina RNA-seq data were aligned and assembled using an in-house pipeline from National Bioinformatics Infrastructure Sweden (NBIS) (https://github.com/NBISweden/pipelines-nextflow/tree/master/subworkflows/transcript_assembly) that includes fastq preprocessing in fastp v. 0.23.2 (Chen et al. 2018), alignment of reads to the rehttps://www.doi.org/10.5281/zenodo.3552717ference genome using HISAT v. 2.1.0 (Kim et al. 2015) and transcript assembly using StringTie v. 2.2.1 (Pertea et al. 2015).
The final assembly was annotated using the MAKER pipeline v.3.01.02 (Holt and Yandell 2011). First, evidence-based-annotations were constructed using both Illumina RNA-seq and Iso-seq assembled transcripts and Uni-Prot reference proteins. Second, using the initial MAKER evidence file, ab initio training was performed with Augustus v.3.3.3 (Stanke et al. 2006) using an NBIS in-house pipeline (https://github.com/NBISweden/pipelines-nextflow/tree/master/subworkflows/abinitio_training).
After running MAKER on a combination of evidence annotation together with curated ab initio profiles, a statistical evaluation of the final annotation was performed using the script agat_sp_statistics.pl from the AGAT package v.0.8.0 (Dainat 2021) followed by complimenting the final evidence-based annotation with the ab initio genes missing in the evidence-based build with agat_sp_complement_annotations.pl from the same toolkit.
Using BLAST v. 2.9.0 (Altschul et al. 1990), searching against the Uniprot/Swissprot databases, with a maximum e-value cutoff of 1e-6, and Interproscan v. 5.59–91.0 (Hunter et al. 2012) functional annotation was performed on the translated CDS features of each coding transcript using the NBIS in house pipeline functional_annotation (https://github.com/NBISweden/pipelines-nextflow/tree/master/subworkflows/functional_annotation). Furthermore, tRNAs were predicted using tRNAscan-SE v.1.3.1 (Lowe and Eddy 1997) and other ncRNAs were predicted using curated Rfam co-variance models (Nawrocki et al. 2014) with the infernal package v.1.1.2 (Nawrocki and Eddy 2013).
Results and discussion
From an adult male B. viridis (Supplementary Fig. S1), we generated 149.53 Gb PacBio HiFi, 468.78 Gb Omni-C and 12.65 Gb Iso-Seq sequencing reads together with 32.56 Gb Illumina RNA-seq data from 7 tissues (Supplementary Table S1). Using Genomescope2 with a k-mer length of 31, the predicted genome size was ∼3.9 Gb with 0.27% heterozygosity (Supplementary Fig. S2). After purging of the initial HiFi assembly (Supplementary Fig. S3 and Table S2), we obtained a completeness score of 99.97 and QV score of 52.6 from MERQURY.FK (Supplementary Table S3). The final scaffolded assembly had a total size of ∼3.89 Gb and consists of 2,107 scaffolds (Supplementary Table S4). The longest scaffold is ∼644 Mb and the assembly has a scaffold N50 of ∼478 Mb (Table 1), with 43% GC content (Supplementary Table S2). Eleven of the 2,107 scaffolds were considerably larger than the others (Supplementary Fig. S4) suggesting that they correspond to the 11 chromosomes. This is in accordance with other published true toad genome assemblies (Bufo gargarizans: Lu et al. 2021, Bufo bufo: Streicher et al. 2021).
BUSCO scores using the tetrapoda_odb10 dataset containing 5,310 genes, for the final scaffolded assembly results in 91.3% complete tetrapod BUSCOs, out of which 90.4% are complete single copy and 0.9% complete duplicated BUSCOs (Supplementary Table S5).
A total of 46.7% (∼1.81 Gb) of the genome was repeat masked with the combined repeat annotations from RepeatModeler v.2.0.2a (Smit and Hubley 2010; Flynn et al. 2020) and RepeatRunner (Yandell 2006) (Supplementary Table S7). The majority of the repeats were classified as unknownl the rest consisted mostly of simple repeats, DNA transposons, LINEs, and LTRs (Table 1). This distribution of repeat families agrees with the expected distribution in Anuran genomes (Zuo et al. 2023).
The initial run of the MAKER software failed for scaffolds (chromosomes) 1, 4, and 11 using the species-specific repeat library for the toad. For those chromosomes, the built-in repeat library in MAKER was instead used in separate runs, and the 2 runs with the species-specific and default repeat library were then merged. The relative repeat densities across chromosomes 1, 4, and 11 suggest that the reason behind this might be that the species-specific repeat identification and masking for those chromosomes might not have been entirely optimal together with the large size of the chromosomes (Fig. 1a).
a) The 11 putative chromosomes order by size with gene density represented as an overlaid heatmap and density of repeats in orange on the right side of each chromosome RIdeogram v.0.2.2 (Hao et al. 2020). The largest chromosomes have a total length of 644 Mb. b) snail plot produced in BlobToolKit (Challis et al. 2020) showing assembly statistics and BUSCO scores for the final scaffolded assembly.
The structure of a total of 31,608 protein-coding genes was annotated, with an average exon and intron length of 310 and 4,984 bp, respectively (Table 1). The annotated gene set covers ∼87% of complete tetrapod BUSCOs (Supplementary Table S6). Due to ab initio predictions showing elevated numbers of duplicated genes and fewer mRNAs, this final annotation was built based on evidence facilitated by Illumina RNA-seq and 219,633 full length cDNAs from Iso-Seq and complemented with ab initio predictions for loci that was missing in the initial evidence-based annotation. Furthermore, an elevated number of single exon genes without annotation was found and subsequently removed in the final build. In total 31,356 genes and 68,100 mRNAs could be linked to functional and protein domains (Supplementary Table S8), out of which 23,830 genes and 56,974 mRNAs could be assigned a name (e-value < 1e-6) by BLAST searches against the Uniprot/Swissprot database. Furthermore, a total of 5,922 tRNAs and 6,112 exons were predicted by tRNAscan-SE v.1.3.1 (Lowe and Eddy 1997) with a total exon length of 434 Kb. However, only 23 tRNAs remained after discarding annotations with an annotation edit distance of 1. The ∼66 Kb candidate sex determination locus for male-heterogametic XY found in Kuhl et al. 2024, consisting of the gene bod1 l (biorientation of chromosomes in cell division 1 was annotated and located in the region chr1: 579239124-579305844.
Finally, comparing the B. viridis assembly to the assemblies of the common toad (Bufo viridis, NCBI: GCF_905171765.1) and the Asiatic toad (B. gargarizans: GCF_014858855.1) reveal that while the genome size for B. viridis is smaller than the other 2 species, the contiguity (scaffold N50, Supplementary Table S9) is lower compared to both other assemblies but comparable to the Asiatic toad assembly. This is a result of the final assembly containing a relatively large number of unplaced scaffolds, the majority of which (67%) are under 100 Kb. It should also be noted that the 2 Bufo assemblies utilizes additional data for scaffolding (10X Genomics Chromium and BioNano data).
Supplementary Material
jkaf002_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Open 2C; Abdennur N, Fudenberg G, Flyamer IM, Galitsyna AA, Goloborodko A, Imakaev M, Venev SV. 2023. Pairtools: From sequencing data to chromosome contacts [Preprint]. bio Rxiv 2023.02.13.528389. 10.1101/2023.02.13.528389.PMC 1116436038809952 · doi ↗ · pubmed ↗
- 2Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. doi:10.1016/S 0022-2836(05)80360-2.2231712 · doi ↗ · pubmed ↗
- 3Challis R, Richards E, Rajan J, Cochrane G, Blaxter M. 2020. Blob Tool Kit—interactive quality assessment of genome assemblies. G 3 (Bethesda). 10(4):1361–1374. doi:10.1534/g 3.119.400908.32071071 PMC 7144090 · doi ↗ · pubmed ↗
- 4Chen S, Zhou Y, Chen Y, Gu J. 2018. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i 884–i 890. doi:10.1093/bioinformatics/bty 560.30423086 PMC 6129281 · doi ↗ · pubmed ↗
- 5Dufresnes C, Mazepa G, Jablonski D, Caliari Oliveira R, Wenseleers T, Shabanov DA, Auer M, Ernst R, Koch C, Ramírez-Chaves HE, et al 2019. Fifteen shades of green: The evolution of Bufotes toads revisited. Mol Phylogenet Evol. 141:106615. doi:10.1016/j.ympev.2019.106615.31520778 · doi ↗ · pubmed ↗
- 6Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Lieberman Aiden E. 2016. Juicebox provides a visualization system for hi-C contact maps with unlimited zoom. Cell Syst. 3(1):99–101. doi:10.1016/j.cels.2015.07.012.27467250 PMC 5596920 · doi ↗ · pubmed ↗
- 7Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. 2020. Repeat Modeler 2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci U S A. 117(17):9451–9457. doi:10.1073/pnas.1921046117.32300014 PMC 7196820 · doi ↗ · pubmed ↗
- 8Formenti G, Theissinger K, Fernandes C, Bista I, Bombarely A, Bleidorn C, Ciofi C, Crottini A, Godoy JA, Höglund J, et al 2022. The era of reference genomes in conservation genomics. Trends Ecol Evol. 37(3):197–202. doi:10.1016/j.tree.2021.11.008.35086739 PMC 13065249 · doi ↗ · pubmed ↗