Phenotypic characterization of a female patient with retinitis pigmentosa caused by a homozygous X-linked RPGRORF15 mutation
Marlene Saßmannshausen, Elisa A. Mahler, Sandrine H. Künzel, Constanze L. Kochs, Frank G. Holz, David Rosenkranz, Hanno J. Bolz, Philipp Herrmann

TL;DR
A female patient with a rare X-linked mutation causing retinitis pigmentosa is described, showing severe retinal degeneration similar to affected males.
Contribution
The paper presents a novel clinical case of a homozygous RPGRORF15 mutation in a female patient with RP.
Findings
The patient exhibited severe retinal degeneration with global photoreceptor loss and decreased FAF signal.
The patient's phenotype was similar to that of her affected uncle, including central photoreceptor residuals and patchy FAF patterns.
The patient's homozygous RPGRORF15 mutation suggests potential eligibility for future therapeutic trials.
Abstract
To describe a detailed phenotypic expression of a homozygous female with retinitis pigmentosa (RP) within a consanguineous family revealing an extremely rare genetic constellation with possible implications for future emerging therapies in addressing inherited retinal dystrophies. Multimodal retinal imaging including wide field fundus photography, fundus autofluoresence (FAF), high-resolution spectral domain optical coherence tomography (SD-OCT) imaging, functional testing comprising visual fields and electroretinogram as well as genetic testing were performed in two consanguine cases of RP. A 44-year-old female patient was referred for evaluation and counseling for potential treatment options presenting with night blindness and visual field defects since early childhood. Extended ophthalmologic examination including multimodal retinal imaging and functional testing showed a clinical…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
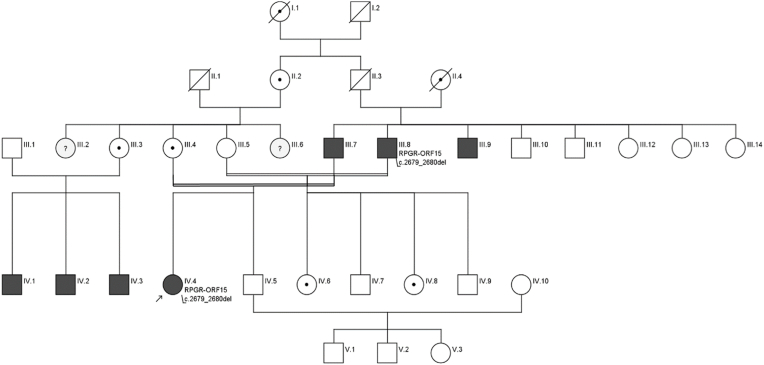 Figure 1
Figure 1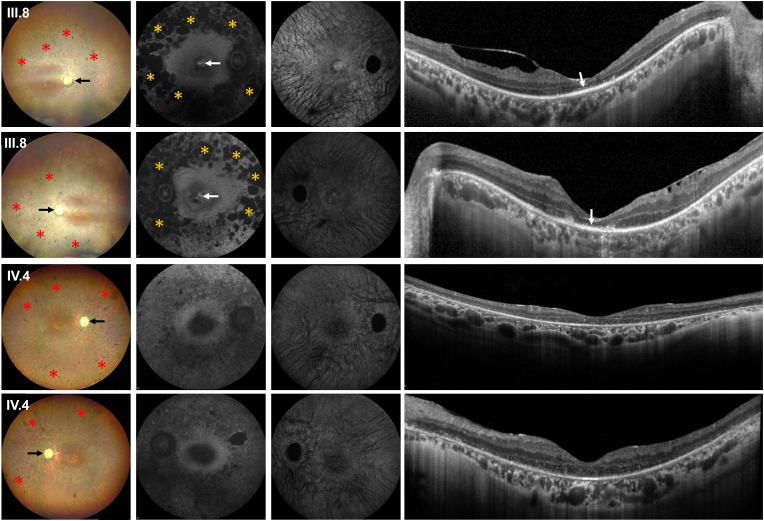 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRetinal Development and Disorders · Retinal Diseases and Treatments · Retinopathy of Prematurity Studies
Introduction
1
Inherited retinal diseases (IRD) are a phenotypically and genetically heterogeneous group of monogenic diseases, which can lead to blindness and visual impairment at an early age. The most common IRD subgroup is retinitis pigmentosa (RP) with an estimated prevalence of up to 1:3000 that is inherited as either autosomal-dominant (30–40 %), autosomal-recessive (50–60 %) or also X-linked (5–15 %).1^,^2 Up to 80 % of the X-linked retinitis pigmentosa (XLRP) cases are linked to mutations detected on the RPGR gene.3, 4, 5, 6, 7 RPGR encodes for the RP GTPase Regulator protein (RPGR), which is responsible for the ciliary transport of metabolites between inner and outer photoreceptor segments.8 Mutations in exons 1–14 account for only 25 % of all disease causing mutations, while the large exon open reading frame 15 (ORF15) harbors up to 80 % of disease-causing RPGR mutations.3 Due to its repetitive purine-rich sequence, genetic testing has been challenging, but different strategies can be applied to detect mutations in ORF15.9
Novel therapeutic approaches like gene augmentation therapy are aiming to slow disease progression, or even to revert the pathological phenotype to some extent. In this context, official clinical approval of Luxturna (voretigene neparvovec) for a single gene causing RP (RPE65) by the Federal Food and Drug Administration in 2017 has given new hopes also for other IRDs. The currently four registered phase III clinical trials are further demonstrating the growing interest in developing treatment for RPGR-related RP (clinicaltrials.gov.: NCT04671433, NCT04794101, NCT03584165, NCT04850118).3
The phenotypical presentation of RPGR-related IRD is broad and comprises a classical rod-cone RP phenotype with early onset of night blindness and difficulties with dark adaptation resulting from mutations most commonly found in exons 1–14.5^,^10 However, mutations in RPGR can also lead to cone-rod dystrophy reported in around 20 % of patients with day-time vision problems, photophobia, dyschromatopisa and blurred central vision.5^,^8^,^10^,^11
Representing an X-linked disease, most patients are hemizygous males while the clinical presentation of female carriers varies considerably.6^,^7^,^12 Females carriers present a spectrum of retinal disease severity, ranging from mild or subclinical retinal phenotype to severe retinal degeneration and significant visual impairment.7
Herein, we demonstrate a comprehensive multimodal retinal imaging-based clinical phenotyping of a female patient from a consanguineous Turkish family with a homozygous pathogenic RPGR variant.
Case report
2
Detailed patients’ characteristics including the identified mutation as well as the corresponding family pedigree are provided in Table 1 and Fig. 1.Table 1. Patients’ characteristics.Table 1IDAgeGenderRefraction [dpt] (sph/cyl) OD/OSBCVA OD/OSGeneZygosityNucleotideClassProteinReferenceIII.850m+1.00/-0.75FCRPGR^ORF15^hemi.c.2679_2680del5p. (Glu894Glyfs184)[14^,^18]+1.50/-1.7520/200IV.444f+0.75/-0.7520/200RPGR*^ORF15^homo.c.2679_2680del5p. (Glu894Glyfs*184)[14^,^18]+0.75/-0.7520/200Abbreviations: f: female, m: male, OD: right eye, OS: left eye, BCVA: best-corrected visual acuity, FC: finger-counting, homo.: homozygous, hemi.: hemizygous.Fig. 1. Family pedigree.Fig. 1. Fig. 2Clinical presentation of patientsFig. 2: Clinical phenotype of the right and left eye of the 50-year-old uncle (III.8) with the hemizygous RPGR^ORF15^ and the 44-year-old female patient (IV.4) with the homozygous RPGR^ORF15^ mutation. In both patients multimodal retinal imaging reveals typical RP characteristics including retinal pigmentary changes in form of bone-spicule and pigment clumpings in the midperiphery (red asterisks) and a waxy pallor of the optic disc (black arrows) in wide-field color fundus photography (first column). The 55° blue-fundus-autofluorescence (FAF, second column) imaging is globally reduced. In patient III.8 there is a paracentral ring of increased FAF with a patchy-like reduced FAF in the mid-periphery of the retina (yellow asterisks) and a well-delineable hyperautofluorescent ring at the foveal center (white arrows). The 55° near-infrared autofluorescence imaging (third column) appears overall similarly reduced in both patients, while horizontal spectral-domain optical coherence tomography (SD-OCT) imaging (last column) reveals a global loss of the photoreceptor layers with evidence of single residuals of photoreceptor cells at the foveal center in patient III.8 (white arrows). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)Fig. 2
A 50-year-old male person (III.8, pedigree) reported night blindness since childhood at the age of 10 and the diagnosis of RP was made at the age of 25, when he noticed a progressive visual impairment with loss of contrast vision and increasing reading problems. At his initial presentation in our IRD clinic, vision was counting fingers and 20/200, residual visual fields were smaller than 5° and photopic and scotopic responses on full-field electroretinogram (ERG) were extinguished. Clinical examination revealed a clinical RP phenotype with retinal pigmentary changes, a paracentral ring of increased FAF with a patchy-like reduced FAF in the mid-periphery of the retina and a well-delineable hyperautofluorescent ring at the foveal center. Spectral domain optical coherence tomography (SD-OCT) imaging demonstrated single centrally remaining photoreceptors. This patient is the paternal uncle of a homozygous female who was referred to our clinic after her initial presentation.
The 44-year-old female patient (IV.4, pedigree) was referred for an extended ophthalmological workup to our IRD service reporting about night blindness and visual field defects since early childhood. At the age of 11 years, she had been diagnosed with RP in Turkey. She further reported progressive visual field loss over the last 10 years. No syndromic findings were reported. Several of her relatives (mostly living in Turkey) have also been diagnosed with RP in the past. Her parents were first-degree cousins. With best-corrected visual acuity (BCVA) of 20/200 in both eyes, clinical examination revealed typical RP characteristics including bilateral posterior subcapsular cataract, retinal pigmentary changes, decreased FAF in the mid-periphery of the retina as well as a global loss of the photoreceptor bands in SD-OCT imaging.7 Her functional testing also revealed outer visual field borders of less than 5° in visual field testing and extinguished photopic and scotopic responses on ERG testing.
Genetic testing was performed by target enrichment with IDT xGen® Exome Research Panel V2.0 (IDT Integrated Technologies, Coraville, IA, USA) using additional RPGR^ORF15^-specfic xGen Lockdown Probes (IDT Integrated Technologies, Coraville, IA, USA), sequencing on Illumina NextSeq500 system (Illumina, San Diego, C, USA), quality-based data filtering incl. sequence read trimming (in house software qlip V1.2) and data evaluation with the SeqNext module software (V5.3.4 JSI Medical Systems, Ettenheim, Germany).13 Performed genetic testing was non-CLIA (Clinical Laboratory Improvements Amendments) certified. Molecular testing revealed a hemizygous status in the male patient for a previously described pathogenic frameshift variant (c.2679_2680del, p. Glu894Glyfs∗184, class 5, RefSeq Number: NM_001034853.2) in ORF15 of the RPGR gene resulting in a stop codon after the inclusion of 183 unrelated residues.14 Our female patient presented the same variant homozygously.
Discussion
3
In this report we present a detailed multimodal retinal imaging-based characterization of a very rare homozygous RPGR^ORF15^ variant detected in a female RP patient with a clinically similar RP disease severity compared to the patient's paternal uncle revealing a hemizygous variant within a consanguine family. The presented homozygous female case highlights the effect of the total absence of any functional protein being produced due to the homozygous gene mutation and therefore needs to be regarded in contrast to heterozygous RPGR female carrier presenting with a remaining wildtype allele and varying clinical extent of X-chromosome inactivation.12^,^15
In X-linked RP, female carriers may typically present with a heterogenous clinical phenotype ranging from being unaffected, mildly affected without functional significance or being severely affected. Apart from the effect of different variants, this wide range of clinical presentation can be explained by the varying extent of X-chromosome inactivation.15 In our female patient, with a homozygous mutation in RPGR, X-inactivation does not play a role as both alleles are equally affected and no functional protein is being expressed. This also explains the severe phenotype of our female patient that is similar to male patients with a hemizygous mutation in RPGR.
While an X-linked homozygous RPGR^ORF15^ variant has previously been described in an Iranian consanguineous family with RP,16 our report for the first time demonstrates a very detailed multimodal retinal imaging phenotyping as well as genotypic correlation in a female homozygous patient. Especially with regards to upcoming genetic therapies in RPGR patients, homozygous RPGR female carriers might potentially benefit from gene augmentation therapy treatments as well. However, additional studies are needed here to prove this claim in further detail. To address this, detailed phenotype to genotype correlations are crucial, not just to characterize the disease, but also to counsel patients also with regards to their own families and children and finally also to identify potential candidates for upcoming gene therapy based therapeutic approaches with special regards to current ongoing trials on RPGR gene augmentation therapy. For female carriers, consideration of the ratio of X-chromosome inactivation and their correlation to clinical disease severity is currently under discussion.12^,^15^,^17 This ratio can help to predict long-term disease severity but might also help to identify future candidates for an early disease intervention, who potentially profit most effectively from future therapies.
To conclude, this case study emphasizes the need of a refined phenotype to genotyping correlation in IRD affected patients to enable an individualized counseling of patients and families as well as to identify potential candidates for future upcoming therapeutic approaches at an early stage of disease.
CRediT authorship contribution statement
Marlene Saßmannshausen: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Elisa A. Mahler: Writing – review & editing, Data curation. Sandrine H. Künzel: Writing – original draft. Constanze L. Kochs: Investigation. Frank G. Holz: Writing – review & editing, Supervision, Funding acquisition, Conceptualization. David Rosenkranz: Writing – review & editing, Investigation. Hanno J. Bolz: Writing – review & editing, Methodology, Investigation. Philipp Herrmann: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization.
Patient consent
Written consent has been obtained from all patients.
Authorship
All authors attest that they meet the current ICMJE criteria for authorship.
Funding
N/A.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Menghini M.Cehajic-Kapetanovic J.Mac Laren R.E.Monitoring progression of retinitis pigmentosa: current recommendations and recent advances Expert opinion on orphan drugs 8202067783223188910.1080/21678707.2020.1735352 PMC 7104334 · doi ↗ · pubmed ↗
- 2Dobyns W.B.Filauro A.Tomson B.N.Inheritance of most X-linked traits is not dominant or recessive, just X-linked Am J Med Genet Part A 129A 20041361431531697810.1002/ajmg.a.30123 · doi ↗ · pubmed ↗
- 3La Martinez-Fernandez de Camara C.Cehajic-Kapetanovic J.Mac Laren R.E.Emerging gene therapy products for RPGR-associated X-linked retinitis pigmentosa Expet Opin Emerg Drugs 27202243144310.1080/14728214.2022.215200336562395 · doi ↗ · pubmed ↗
- 4Branham K.Othman M.Brumm M.Mutations in RPGR and RP 2 account for 15% of males with simplex retinal degenerative disease Investig Ophthalmol Vis Sci 532012823282372315061210.1167/iovs.12-11025 PMC 3522443 · doi ↗ · pubmed ↗
- 5Talib M.van Schooneveld M.J.Thiadens A.A.Clinical and genetic characteristics of male patients with rpgr-associated retinal dystrophies: a long-term follow-up study Retina 392019118611992952897810.1097/IAE.0000000000002125 · doi ↗ · pubmed ↗
- 6Bader I.Brandau O.Achatz H.X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF 15Investig Ophthalmol Vis Sci 442003145814631265757910.1167/iovs.02-0605 · doi ↗ · pubmed ↗
- 7Nanda A.Salvetti A.P.Clouston P.Downes S.M.Mac Laren R.E.Exploring the variable phenotypes of RPGR carrier females in assessing their potential for retinal gene therapy Genes 9201810.3390/genes 9120643 PMC 631636930567410 · doi ↗ · pubmed ↗
- 8Megaw R.D.Soares D.C.Wright A.F.RPGR: its role in photoreceptor physiology, human disease, and future therapies Exp Eye Res 138201532412609327510.1016/j.exer.2015.06.007PMC 4553903 · doi ↗ · pubmed ↗