Six Chinese patients with propionic acidemia: from asymptomatic to death in the neonatal period
Shunan Wang, Lulu Li, Yulan Ma, Haihe Yang, Yuting Sang, Yue Tang, Lifei Gong, Jinqi Zhao, Lijin Gu, Yuanyuan Kong, Xinmei Mao

TL;DR
This study reports six Chinese patients with propionic acidemia, a rare metabolic disorder, and identifies new genetic mutations linked to different disease severities.
Contribution
The study expands the genetic mutation spectrum of propionic acidemia and provides insights into genotype-phenotype correlations in Chinese patients.
Findings
The incidence of propionic acidemia was 1 in 114,820 in Beijing and 1 in 189,671 in Ningxia.
Five novel mutations in PCCA and PCCB genes were identified, including one in PCCA and four in PCCB.
Certain mutations were associated with late-onset or early-onset disease progression.
Abstract
Propionic acidemia (PA) is a severe organic acidemia that can result in multi-organ damage and is potentially fatal. The rarity of this disease and the limited number of reported cases contribute to a lack of comprehensive knowledge, particularly concerning the genotype-phenotype correlation. This study aims to report on PA cases in Beijing and Ningxia, China, identify the pathogenic genetic factors involved, and explore the relationship between genotype and phenotype. We calculated the positive screening rates of PA in Beijing and Ningxia and summarized data from six Chinese patients with PA identified at the Beijing Newborn Screening Center and Ningxia Newborn Screening Center. Clinical examinations included blood tandem mass spectrometry, urine gas chromatography-mass spectrometry, and the next-generation sequencing (NGS) technology. Candidate mutations were validated using…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
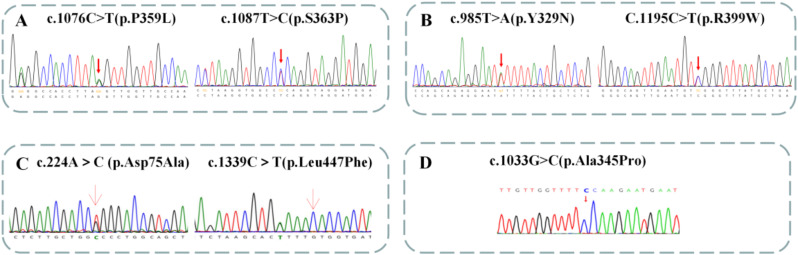 Figure 1
Figure 1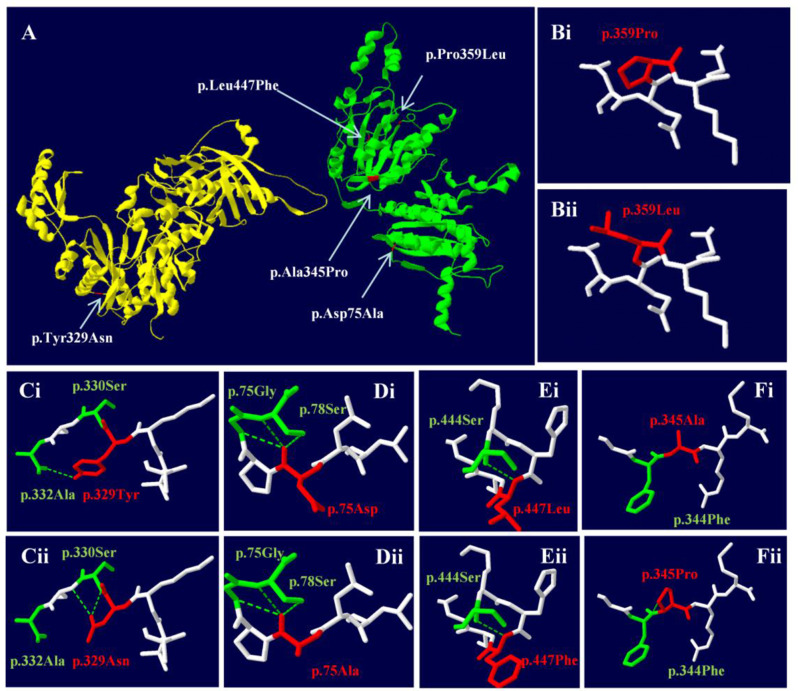 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetabolism and Genetic Disorders · Mitochondrial Function and Pathology · Folate and B Vitamins Research
Introduction
Propionic acidemia (PA) is an organic acid disorder caused by mutations in the PCCA or PCCB genes [1]. These mutations impair propionyl coenzyme A carboxylase (PCC), leading to metabolic disorders involving propionate, propiogenic amino acids, and odd-chain fatty acids [2]. As a result, abnormal metabolites such as methylcitrate, 3-hydroxypropionic acid, and propionylglycine accumulate, causing damage to multiple organs, including the heart, brain, liver, kidneys, bone marrow and pancreas, and can be fatal [3–9]. The symptoms of PA are heterogeneous, ranging from asymptomatic to severe, depending on the degree of enzyme deficiency [10]. PA is classified as early-onset (within the first 3 months of life) and late-onset (after 3 months) [10, 11]. Previous studies have shown that early-onset PA is associated with higher mortality, whereas late-onset PA has a better prognosis, such as fewer frequencies of metabolic disorders and less neurocognitive impairment [12]. The primary treatment for PA is dietary management, which restricts propiogenic amino acids, propionate, and odd-chain fatty acids. If dietary management is insufficient to prevent severe clinical manifestations, liver transplantation is the next option [1]. The prevalence of PA varies globally. In most regions, the incidence of PA is below 1 per 100,000 newborns, except in the Middle East and North Africa [13]. In northern China, the prevalence of PA ranges from 1 in 283,459 to 1 in 3,156 [14].
In this study, we investigated six Chinese patients diagnosed at the Beijing Newborn Disease Screening Center and the Peking University First Hospital Ningxia Women and Children’s Hospital (Ningxia Hui Autonomous Region Maternal and Child Health Hospital). Five patients were diagnosed through newborn screening (NBS), and one was clinically confirmed. We analyzed their diagnostic process, treatment, and prognosis, as well as their biochemical and genetic features. Additionally, we reported the updated prevalence of PA in Beijing and Ningxia. The aim of this study was to broaden the spectrum of clinical features and gene mutations in patients with PA and to explore the relationship between phenotype and genotype in these patients.
Materials and methods
Newborn screening
In accordance with the “Technical Specification for Newborn Screening (2010 Edition),” heel blood was collected from newborns who had been breastfed for 72 h after birth. The samples were dried on S&S903 blood filter paper and then sent to the Beijing Newborn Screening Centre or the Ningxia Newborn Screening Centre for testing. Blood samples from 344,461 neonates in Beijing (collected between April 2014 and March 2024) and 379,341 neonates in Ningxia (collected between May 2016 and March 2024) were measured for amino acids and acylcarnitines levels using tandem mass spectrometry (MS/MS). Elevated propionylcarnitine (C3) and its ratio to acetylcarnitine (C2), free carnitine (C0), and methionine (Met) will prompt a recall for further review.
Patients clinical features and ethics statement
The study protocol was reviewed and approved by the Medical Ethics Committee of Beijing Maternity Hospital of Capital Medical University (2022-KY-087-01). All of the guardians voluntarily participated in the study and signed informed consent forms. We studied six patients: five were diagnosed through newborn screening (three from Beijing and two from Ningxia) and one through clinical onset (from Ningxia). Among them, five were male and one was female.
Metabolic examinations
Neonatal screening for amino acid and carnitine spectrum was conducted using tandem mass spectrometry (TQ Acquity Mass Spectrometer, Waters Corporation, USA). Clinical testing for amino acid and carnitine spectrum was performed using the NeoBase Non-derivatized MSMS Kit (PerkinElmer, Turku, Finland). For differential diagnosis of other metabolic diseases, urine organic acids were tested by gas chromatography-mass spectrometry (Shimadzu GCMS-QP2010).
Genetic testing and bioinformatics analysis
High-throughput sequencing was performed for genetic testing. Briefly, 2 ml of blood was extracted from the proband and the parents. DNA was sequenced using the Illumina HiSeq 2500 system (Illumina, San Diego, USA). After filtering the raw data, clean data were aligned to the human genome reference sequence hg19. Mutations were compared with databases such as HGMD, gnomAD, and ExAC.
Predictive analysis of pathogenicity
Functional prediction of missense mutations was performed by PolyPhen-2, MutationTaster, Sorting Intolerant from Tolerant (SIFT), and REVEL. Variant frequencies were determined using the 1000 Genomes Project, ExAC, and gnomAD databases. The variants were interpreted following the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines.
Swiss-PDB Viewer software was used to predict and evaluate the crystal structure of the mutant proteins. The protein structures of PCCA and PCCB (PDB ID: 7YBU) were acquired from the PDB database and analyzed using Swiss-PDB Viewer 4.1.0 to visualize the protein structures and predict the effects of mutation sites on the tertiary protein structures.
Results
The prevalence of PA in Beijing and Ningxia
In Beijing, 344,461 neonates were tested between April 2014 and March 2024. Three patients were diagnosed with PA (Table 1), resulting in an overall incidence of 1/114,820. In Ningxia, 379,341 neonates were tested between May 2016 and March 2024. Two patients were diagnosed with PA (Table 1), with an overall incidence of 1/189,671.
Table 1. Basic information of the patients with PACaseGenderBirth weightDiagnosis by NBSAge of onsetTargeted treatmentClinical outcomeClinical phenotype1Female3000Yes1dNoDied at 4dEarly-onset2Male2370Yes-NoNormal neurodevelopmentLate-onset3Male3190Yes-YesNormal neurodevelopmentLate-onset4Male3150Yes3 mDiscontinued at 3 mDied at 3 mEarly-onset5Male3500No20dNoDied at 40dEarly-onset6Male4300Yes16dYesDied at 18 mEarly-onset
Clinical features of six patients with PA
Case 1
A female, full-term newborn (40 weeks, BW 3000 g) presented with coma and intermittent convulsions at 1 day old. Besides neonatal convulsions, she had complications including myocardial damage, renal insufficiency, hypocalcemia, neonatal hyperglycemia, neonatal jaundice, and hyperammonemia. She died at 4 days old. She was eventually diagnosed with PA. Blood for newborn screening was collected at 3 days old, and she was registered at the Beijing Newborn Screening Center for “elevated C3, C3-related ratios, and several amino acids” at 15 days old. The final diagnosis was confirmed as PA.
Case 2
A male, preterm newborn (36 weeks, BW 2370 g) was registered at the Beijing Newborn Screening Center at 18 days old for “elevated C3/C2 and C3/C0 on neonatal disease screening.” Repeat findings confirmed significant increases in C3 and C3/C2. He was diagnosed with PA by genetic testing, but the parents refused treatment and follow-up. The last follow-up was conducted by telephone when he was 17 months old, His weight was 12 kg (50-75th percentile) and his length was 82 cm (50th percentile). He was able to say “mama”, “dada”, and some common object names, climb stairs, and follow simple commands.
Case 3
A male, full-term newborn (39 weeks, BW 3190 g) was registered at the Beijing Newborn Screening Center at 20 days old for “elevated C3/C2 and decreased citrulline on neonatal disease screening.” Repeat results indicated mildly elevated C3 and C3/C2. He was diagnosed with PA and started dietary treatment at 41 days old. Starting at 2 months old, his creatine kinase (CK) and creatine kinase-MB (CK-MB) were continuously twice the upper limit of normal, indicating potential myocardial injury. He started treatment with Coenzyme Q10 (10 mg/d), fructose dipllosphate sodium (1 g/d), and levocarnitine (50 mg/kg/d). When he was 3 months old, he conducted his electrocardiogram and cardiac ultrasound, and the results were normal. The last follow-up, conducted at 10 months of age, showed his weight was 12.5 kg (> 97th percentile) and his length was 80 centimeters (> 97th percentile). His intellectual assessment results were normal, as measured by the 0–6 Years Children’s Neuropsychological Development Scale. His CK, CK-MB, electrocardiogram and cardiac ultrasound were all within normal limits. His treatment regimen included dietary management (with the ratio of special treatment milk powder to ordinary milk powder ranging from 1:2 to 1:3), coenzyme Q10 (25 mg/d), and levocarnitine (210 mg/kg/d).
Case 4
A male, full-term newborn (38 weeks + 4 days, BW 3150 g) was registered at the Ningxia Newborn Screening Center at 27 days old for “elevated C3 and C3/C2 on neonatal disease screening.” He was diagnosed with PA and started on dietary treatment (special treatment milk powder: ordinary milk powder = 1:2) and L-carnitine (100 mg/kg/d) at 1-month-old. After one week of treatment, his blood C3/C2 levels did not significantly change, but the associated metabolites in his urine (3-hydroxypropionic acid, propionylglycine, tiglylglycine, methylcitric acid) were significantly reduced (Tables 2 and 3). At 2 months old, his weight was 5.2 kg (25th percentile) and his length was 58.5 cm (50th percentile). He presented visual tracking and was able to lift his head. However, his treatment was discontinued, and he died at 3 months old.
Table 2. Blood tandem-mass spectrometry results of the patientsCaseAgeBlood tandem mass spectrometry (µmol/L)C3 (0.4−5)C3/C2 (0.03–0.2)C3/C0 (0.02–0.27)C3/MET (0.02–0.2)C0 (8.6–60)MET (10–55)Gly (180–1000)13d10.830.821.570.236.9147.83650.9323d4.110.330.260.2915.8414.01258.41233d17.860.570.530.7834.0233d3.290.260.230.1214.3528.13373.87320d5.30.530.160.1833.1729.6204.4243d18.652.332.661.43713518434d83.072.312.6027.69323782532d17.496.561.2314.2563d21.560.710.970.9222.2923.31838.94611d12.251.241.130.5510.8622.211225.43
Table 3. Gas chromatography-mass spectrometry results of the patientsCaseAgeGas chromatography-mass spectrometry (mmol/mmol Cr)Methylcitric acid (0−0.7)Methylcitric acid−4 (0−0.8)3-Hydroxypropionic acid−2 (0.0–4.0)Propionylglycine (0−0.4)Tiglylglycine−1(0.0–5.0)Tiglylglycine−2(0.0−0.5)320d2.052.67.3200.15032 m1.491.865.5600.180427d39.239.2107.816.517.6156.2434d34.234.229.82.92.40532d8.78.779.715.57.5NA611d29.720.7193.88.734.78.6
Case 5
A male, full-term newborn (BW 3500 g) from a consanguineous marriage presented with drowsiness and feeding difficulties from 24 days old after a fever. He was taken to a local hospital. He had complications of pyemia, protein-energy malnutrition, hypoimmunoglobulinemia, neonatal anemia, thrombocytopenia, atrial septal defect, electrolyte disturbance (hyponatremia, hypocalcemia), and metabolic acidosis with respiratory alkalosis. He died at 40 days old. He was diagnosed with PA by blood and urine metabolic tests.
Case 6
The younger brother of Case 5, a male, full-term newborn (38 weeks + 4 days, BW 4300 g), was registered at the Ningxia Newborn Screening Center at 11 days old for “significantly elevated C3 and related ratios on neonatal disease screening.” Since his brother was suspected to have died of an inherited metabolic disease, he started dietary treatment (special: regular milk powder = 2:1), L-carnitine (250 mg/kg/d), and hydroxocobalamin (1 g/d) at 11 days old before diagnosis. At 16 days old, he showed poor feeding and a lack of mental alertness. His blood ammonia was 196 µmol/l. EEG suggested mildly abnormal activity, and cranial ultrasound indicated slightly enhanced white matter echoes. Cranial MRI suggested abnormal signals in several brain regions. His treatment was adjusted to include arginine (0.9 g/d) and sodium benzoate (0.9 g/d). His clinical symptoms improved, and ammonia levels normalized. At 18 months of age, following a respiratory tract infection, he developed metabolic acidosis again, accompanied by type 1 respiratory failure. Despite active treatment, including acid correction, infection control, and respiratory support, the last arterial blood gas analysis showed a pH of 6.94, PCO2 of 83.3 mmHg, and lactate levels that were too high to measure. He passed away after emergency interventions were discontinued.
Metabolic examinations
All patients except Case 5 were diagnosed with NBS. All five patients showed elevated C3/C2 during NBS. C3 and C3/C0 were variably elevated. All cases showed elevated C3, C3/C2, and C3/C0 upon re-check (Table 2). Except for Case 3, who had elevated 3-hydroxypropionic acid but normal propionylglycine, all other patients exhibited elevated propionylglycine, 3-hydroxypropionic acid, tiglylglycine, and methylcitric acid (Table 3).
Gene sequencing results
Four patients did genetic tests. Next-generation sequencing revealed that one patient carries mutations in the PCCA gene, while three patients carry mutations in the PCCB gene. Specifically, the genetic test identified two mutations in the PCCA gene (c.985T > A and c.1195 C > T) and five mutations in the PCCB gene (c.1076 C > T, c.1087T > C, c.224 A > C, c.1339 C > T, and c.1033G > C). Among these, one novel mutation (c.985T > A) was found in PCCA, and four novel mutations (c.1076 C > T, c.224 A > C, c.1339 C > T and c.1033G > C) were identified in PCCB. For detailed information, please refer to Table 4; Fig. 1.
Table 4. Genetic results of the patients and bioinformatic analysis of novel missense variantsCaseClinical phenotypeGenetic characteristicsExACALLGnomADALLSIFTPolyphen2Mutation TasterREVELACMGGeneAllele originVariant locationNucleotideNovel variantVariant type2Late-onset PCCB PE10c.1076 C > T(p.P359L)YM--D(0)-D(1)D(0.89)VUSME10c.1087T > C(p.S363P)NM3Late-onset PCCA PE12c.985T > A(p.Y329N)YM--D(0)-D(1)D(0.94)LPME13c.1195 C > T(p.R399W)NM4Early-onset PCCB PE2c.224 A> C(p.D75A)YM--D(0)-D(1)D(0.99)LPME13c.1339 C> T(p.L447F)YM--U(0.001)-D(1)D(0.79)P6Early-onset? PCCB P/ME10c.1033G > C(p.A345P)YM--U(0.006)-D(1)D(0.95)LPN: Nonsense; M: Missense. D: Deleterious; U: Uncertain; P: Pathogenic; LP: Likely pathogenic
Fig. 1. Available genetic testing analysis of patients with PA. (A) In case 2, sanger sequencing revealed the patient had the compound heterozygous variants c.1076 C > T (p.P359L) of paternal origin and c.1087T > C (p.S363P) of maternal origin in the PCCB gene. (B) In case 3, sanger sequencing revealed the patient had the compound heterozygous variants c.985T > A (p.Y329N) of paternal origin and c.1195 C > T (p.R399W) of maternal origin in the PCCA gene. (C) In case 4, sanger sequencing revealed the patient had the compound heterozygous variants c.224 A> C (p.Asp75Ala) of paternal origin and c.1339 C> T (p.Leu447Phe) of maternal origin in the PCCB gene. (D) In case 6, the patient had the compound homozygous variant c.1033G > C (p.Ala345Pro) in the PCCB gene
Pathogenicity prediction analysis
Variations c.985T > A in the PCCA gene and c.1076 C > T, c.224 A > C, c.1339 C > T and c.1033G > C in the PCCB gene are novel mutations not reported in the 1000 Genomes Project, gnomAD, or ExAC databases (Table 4). Missense mutations were analyzed using PolyPhen-2, MutationTaster, SIFT, and REVEL. All mutations were consistent with the patient’s clinical phenotype, which supports pathogenic evidence (Table 4).
Swiss-PDB software was used to predict the effect of missense mutations on protein conformation in PCCA and PCCB (Fig. 2A). Mutations c.1076 C > T (p.P359L) (Fig. 2B), c.224 A > C (p.Asp75Ala) (Fig. 2D), and c.1339 C > T (p.Leu447Phe) in PCCB (Fig. 2E) altered the side chain structure of the protein. Mutations c.985T > A in PCCA (Fig. 2C) and c.1033G > C (p.Ala345Pro) in PCCB (Fig. 2F) affected both the side chain structure and hydrogen bond linkage with adjacent amino acids.
Fig. 2. Three-dimension structure of PCCA and PCCB gene (wild type and mutant). (A) The three-dimension structure of the PA protein. The chain αwas highlighted in yellow, and the chain βin green. (B) The mutation changes the side chain structure. (C) The mutation makes it lose one hydrogen bond with alanine 332 and form a hydrogen bond with serine 331, which is expected to affect the stability of the protein structure. (D) The mutation changes the side chain structure. (E) The mutation changes the side chain structure. (F) The mutation leads to an additional hydrogen bond with phenylalanine 344
Discussion
PA is a rare inherited metabolic disorder with high disability and mortality rates. We conducted a clinical and genetic study on six patients, broadening the spectrum of clinical phenotype spectrum of PA and the mutation spectrum of the PCCA and PCCB genes. We also explored the genotype-phenotype relationship in patients with PA. Despite the small sample size, our cases ranged from asymptomatic to death in the neonatal period, making them representative. In addition, we reported the updated incidence of PA in Beijing and Ningxia, China.
In our study, the incidence of PA in Beijing is 1/114,820, and in Ningxia is 1/189,671, which aligns with previously reported rates [15]. The overall incidence of PA in northern China was from 1/283,459 to 1/3,156 [14]. In Beijing, one patient carried PCCB gene mutations and one patient carried PCCA gene mutations. All patients (100%) in Ningxia carried PCCB gene mutations, which is inconsistent with a previous study where 24 (41.4%) patients had PCCA variants and 34 (58.6%) had PCCB variants [10]. This discrepancy may be due to the limited number of cases. We plan to continue studying the prevalence as our sample size increases.
The PCCA gene (OMIM 232000) is located on 13q32.3, contains 24 exons, and encodes 728 amino acids. The PCCB gene (OMIM 232050) is located on 3q22.3, contains 15 exons, and encodes 539 amino acids. They encode the α and β subunits of the mitochondrial biotin-dependent enzyme propionyl-CoA carboxylase, respectively [16]. The α subunit mainly contains three functional domains: the C-terminal biotin binding region, the biotin transfer region, and the N-terminal biotin carboxylase region. The β subunit hexamerizes with a propionyl-CoA binding site and a carboxyltransferase domain responsible for transferring the carboxyl group to propionyl-CoA [14]. Mutations in PCCA or PCCB genes lead to decreased PCC activity, causing PA. PA can be categorized into early-onset (< 3 months) and late-onset (> 3 months) forms [10]. Earlier onset is associated with more severe symptoms and worse prognosis. There is thought to be a connection between genotype and phenotype in PA patients, but further studies are needed [17]. The connection between gene mutation types, distribution, and phenotype remains unclear, but certain mutations may be related to the late-onset form or early-onset form of PA.
In our study, only c.1087T > C (p.S363P) in PCCB and c.1195 C > T (p.R399W) in PCCA were previously reported. The c.1087T > C (p.S363P) mutation in PCCB is related to the late-onset type [10]. The c.1195 C > T (p.R399W) mutation in PCCA is associated with a severe phenotype [18]. Additionally, we reported one novel mutation in the PCCA gene and six novel mutations in the PCCB gene. All these mutations have very low frequencies in the population and were predicted to be pathogenic by software analysis. In China, missense mutations are the most frequent type in PCCA and PCCB. In our study, 5 out of 7 (71.4%) were missense mutations.
The only patient (Case 3) with PCCA gene mutations had a late-onset form. All his blood and urine metabolic markers were only mildly elevated, and he had normal physical and mental development. He carries c.985T > A and c.1195 C > T, both in the biotin carboxylase region of PCCA. The latter is related to a severe phenotype, but given his mild clinical presentation, c.985T > A may be associated with a late-onset phenotype. Although his electrocardiogram and cardiac ultrasound results were normal, his CK and CK-MB levels were mildly elevated from 2 months of age, indicating that his cardiac function requires further follow-up.
Three patients (Cases 2, 4, and 6) carried PCCB gene mutations. Case 2 carries compound heterozygous mutations c.1076 C > T and c.1087T > C (p.S363P) in PCCB. The latter is a common mutation in Chinese PA patients and is related to the late-onset type, consistent with our findings [10]. Given his normal development, the novel mutation c.1076 C > T in PCCB may also be associated with late-onset. After treatment was discontinued, Case 4 died passed away at 3 months of age, suggesting that c.224 A > C (p.Asp75Ala) and c.1339 C > T (p.Leu447Phe) may be associated with early-onset. Case 5 and Case 6 are brothers, hypothesized to have identical genotypes and similar phenotypes. Case 5 had an early-onset form and died at 40 days old. Case 6 developed symptoms at 16 days of age and died at 18 months of age, and was found to carry homozygous mutations c.1033G > C (p.Ala345Pro) in PCCB. Based on the clinical presentation of these two cases, this mutation may be associated with an early-onset phenotype.
Early detection and treatment through newborn screening can improve the short-term prognosis. In our study, after one week of treatment, Case 4’s metabolic indicators improved significantly. His urine propionylglycine, 3-hydroxypropionic acid, methylcitric acid, and tiglylglycine decreased to near-normal levels, though methylcitric acid slightly decreased. His growth and development were normal until the treatment was discontinued. Case 5 and Case 6 are brothers and may carry the same mutations in the PCCA gene. Case 5 presented with drowsiness and feeding difficulties from 24 days old and died at 40 days old. However, his younger brother (Case 6) was called by the NBS center and accepted treatment at 11 days old. He died at 18 months of age. These cases indicates that early diagnosis and treatment can significantly improve short-term outcomes. However, in this study, early diagnosis and treatment through newborn screening did not result in an ideal long-term prognosis for patients with early-onset propionic acidemia. Current treatment mainly involves protein restriction [1], which fails to completely block the pathogenesis of PA or prevent poor prognosis. Some children still develop recurrent metabolic acidosis, cardiomyopathy, seizures, and developmental delays/intellectual disability [19, 20]. Therefore, studies related to longer follow-up and more effective treatments are needed to improve the long-term prognosis of PA patients.
In addition to metabolic disorders and neurological damage, myocardial involvement is a significant complication of PA. It can develop into arrhythmia, cardiomyopathy, severe heart failure, long-QT syndrome, or even cardiac arrest [21], which is life-threatening. Though Case 3 presented with slightly abnormal blood and urine metabolites and normal development, his CK and CK-MB were continuously elevated starting at 2 months old, implying that cardiac damage in PA patients may not be due to metabolic abnormalities alone but may involve other aspects of pathogenesis. His heart function needs long-term monitoring. Case 2 refused treatment. While he has not shown any intellectual developmental issues thus far, vigilance is required for potential cardiac complications or even the risk of sudden cardiac death. Due to the limited time and sample size of the study, we did not observe more complications. We will continue to expand the sample size and study time for further follow-up studies.
Conclusion
The incidence of PA in Beijing is 1/114,820 and in Ningxia is 1/189,671. We reported six patients with PA and identified one novel mutation (c.1195 C > T) in the PCCA gene and four novel mutations (c.1076 C > T, c.224 A > C, c.1339 C > T, and c.1033G > C) in the PCCB gene. Mutations c.985T > A in PCCA and c.1076 C > T in PCCB may be associated with late-onset, while c.224 A > C, c.1339 C > T, and c.1033G > C in PCCB may be associated with early-onset.
Limitation
Firstly, our study is limited by a small sample size, which may impact the robustness of genotype-phenotype correlation analyses. We will expand the sample size in further studies. Secondly, our follow-up time was limited. The long-term prognosis of PA patients with early diagnosis and treatment from neonatal screening remains unclear. We will conduct a longer-term follow-up study. Lastly, we were unable to perform functional experiments in this study. We aim to include these analyses in future research to deepen our understanding of the identified mutations.