Discovery of N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-phenoxyacetamide Derivatives as Novel Potential BCR-ABL1 Inhibitors Through Structure-Based Virtual Screening
Shuaixing Wang, Minyi Wang, Zi Li, Guofeng Xu, Dayan Wang

TL;DR
Researchers discovered new compounds that inhibit the BCR-ABL1 kinase, a key target in treating chronic myeloid leukemia, and showed they work well alone and with existing drugs.
Contribution
A new class of BCR-ABL1 inhibitors was identified through virtual screening and demonstrated anti-tumor activity.
Findings
Compound 10m inhibited BCR-ABL-dependent signaling with an IC50 of 0.98 μM in K562 cells.
Compound 10m synergized with asciminib to enhance anti-proliferation and pro-apoptotic effects.
Molecular dynamics simulations helped identify novel scaffolds with potential as BCR-ABL1 inhibitors.
Abstract
BCR-ABL1 kinase is a critical driver of chronic myeloid leukemia (CML) pathophysiology. The approval of allosteric inhibitor asciminib brings new hope for overcoming drug resistance caused by mutations in the ATP-binding site. To expand the chemical diversity of BCR-ABL1 kinase inhibitors with positive anti-tumor effect with asciminib, structure-based virtual screening and molecular dynamics simulations were employed to discover novel scaffolds. This approach led to the identification of a series of N-(2-acetamidobenzo[d]thiazol-6-yl)-2-phenoxyacetamide derivatives as new BCR-ABL1 inhibitors. The most potent compound, 10m, demonstrated inhibition of BCR-ABL-dependent signaling and showed an anti-tumor effect against K562 cells, with an IC50 value of 0.98 μM. Compound 10m displayed powerful synergistic anti-proliferation and pro-apoptotic effects when combined with asciminib,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1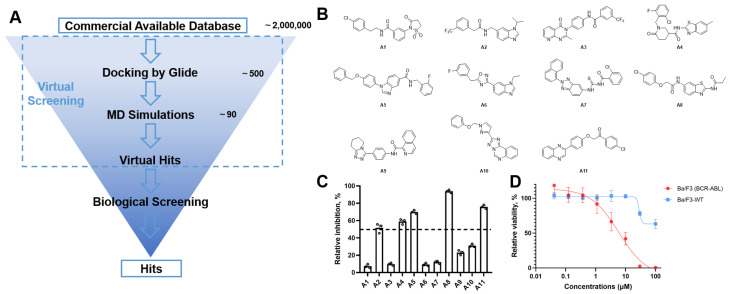 Figure 2
Figure 2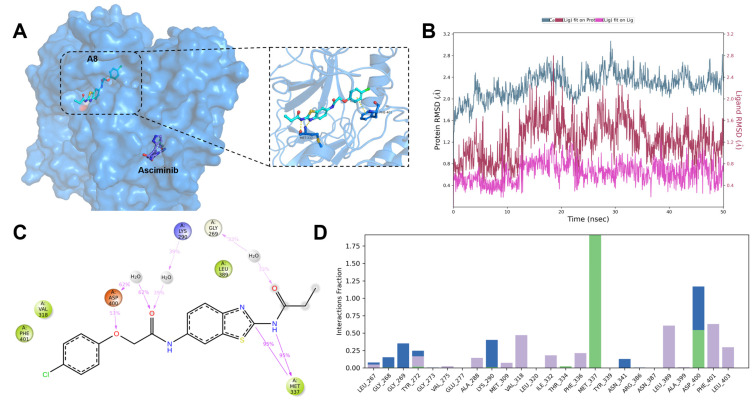 Figure 3
Figure 3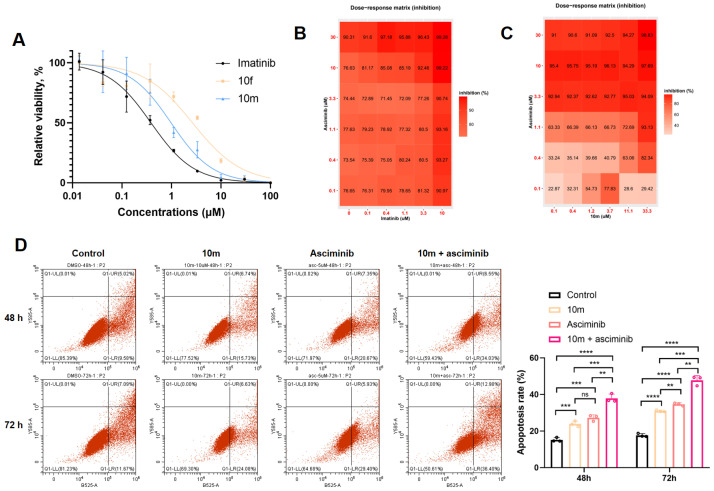 Figure 4
Figure 4 Figure 5
Figure 5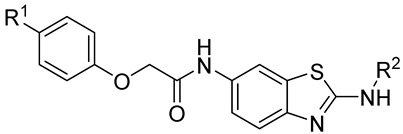 Figure 6
Figure 6 Figure 7
Figure 7 Figure 8
Figure 8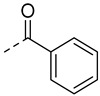 Figure 9
Figure 9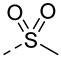 Figure 10
Figure 10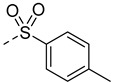 Figure 11
Figure 11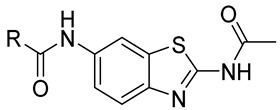 Figure 12
Figure 12 Figure 13
Figure 13 Figure 14
Figure 14 Figure 15
Figure 15 Figure 16
Figure 16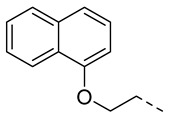 Figure 17
Figure 17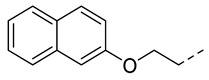 Figure 18
Figure 18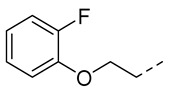 Figure 19
Figure 19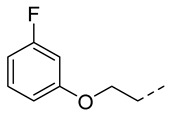 Figure 20
Figure 20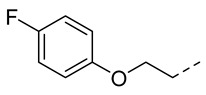 Figure 21
Figure 21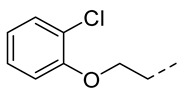 Figure 22
Figure 22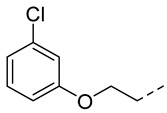 Figure 23
Figure 23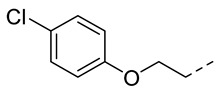 Figure 24
Figure 24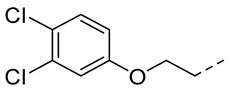 Figure 25
Figure 25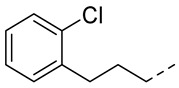 Figure 26
Figure 26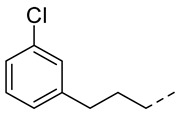 Figure 27
Figure 27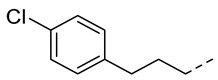 Figure 28
Figure 28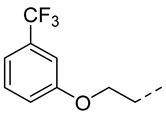 Figure 29
Figure 29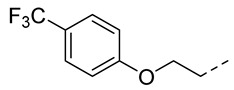 Figure 30
Figure 30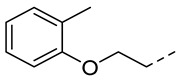 Figure 31
Figure 31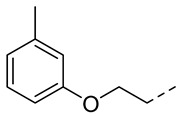 Figure 32
Figure 32 Figure 33
Figure 33 Figure 34
Figure 34 Figure 35
Figure 35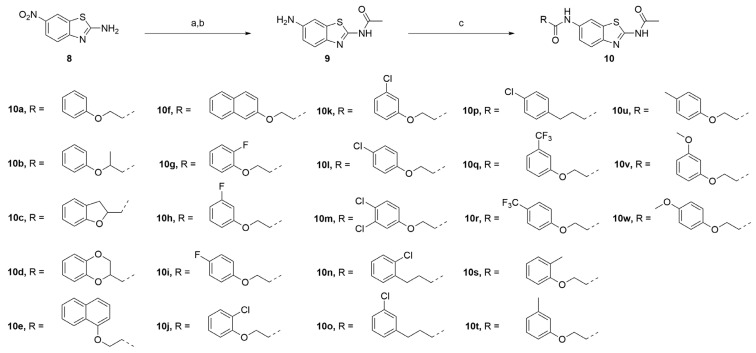 Figure 36
Figure 36- —Young Scholar Science Foundation of China CDC
- —National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChronic Myeloid Leukemia Treatments · Myeloproliferative Neoplasms: Diagnosis and Treatment · Chronic Lymphocytic Leukemia Research
1. Introduction
Chronic myeloid leukemia (CML) is a hematological malignancy characterized by the presence of the Philadelphia chromosome, resulting from a reciprocal translocation between chromosomes 9 and 22, namely t (9;22) (q34;q11) chromosome translocation. This translocation generates the Bcr-Abl1 fusion gene, which encodes a constitutively active tyrosine kinase (BCR-ABL1) that drives aberrant cell proliferation and impaired apoptosis [1,2]. CML accounts for approximately 15% of all adult leukemias and has a global incidence rate of 1–2 cases per 100,000 annually [3]. The BCR–ABL1 kinase drives multiple signaling pathways, including cell growth, survival, invasion and angiogenesis and tumor initiation and progression, indicating poor prognosis in CML patients and acute lymphoblastic leukemia (ALL) patients [4].
The advent of tyrosine kinase inhibitors (Figure 1), notably imatinib (1) [5], has transformed CML into a manageable chronic disease, with most patients achieving near-normal life expectancy when treated during the chronic phase [6]; however, several therapeutic challenges remain. Resistance to treatment is a major concern, particularly in advanced disease stages, which spurred the discovery of second-generation inhibitors, including nilotinib (2) [7], dasatinib (3) [8], bosutinib (4) [9], etc. The T315I “gatekeeper” mutation is among the most refractory, rendering the majority of ATP-competitive TKIs ineffective. Second-generation inhibitors such as dasatinib and nilotinib have improved potency against several resistant mutations; however, their efficacy against T315I and certain other mutations remains limited [10,11]. Third-generation inhibitors like ponatinib (5) [12] and olverembatinib (6) [13] address this gap but are associated with severe toxicity risks, including cardiovascular complications [14,15]. Therapeutic strategies are being pursued to overcome these limitations. Asciminib (ABL001, 7) [16], the first allosteric BCR-ABL1 inhibitor demonstrates activity against TKI-resistant mutations, including T315I, and offers a potentially improved safety profile by minimizing off-target kinase inhibition. However, rapid development of drug resistance also has been observed [17]. One promising approach is the combination therapy dual-targeting the ATP-binding site and myristoyl pocket of BCR-ABL1, which provides an alternative mechanism of action by restoring the protein’s auto-inhibitory conformation. In vitro studies demonstrated additive effects of asciminib when combined with imatinib, nilotinib, or dasatinib [16,17,18]. Asciminib also exhibited anti-tumor activity against BCR-ABL1 point mutants and suppressed the emergence of resistant point mutations when combined with nilotinib or ponatinib [19]. However, negative cooperativity between imatinib and asciminib has been observed in a structural study [20], underscoring the importance of rational selection of double-drug combinations with positive cooperativity.
In this work, structure-based in silico methods were employed to discover an orthosteric inhibitor bearing synergistic anti-tumor potency with asciminib. Hit compound A8 exhibited moderate growth inhibition against Ba/F3 (BCR-ABL1) cells, with a GI_50_ value of 6.4 μM. 23 N-(benzo[d]thiazol-2-yl)acetamide derivatives were designed and synthesized, and their anti-tumor activities against Ba/F3 and K562 cells as well as BCR-ABL1-dependent signaling inhibitory potency were evaluated. The most potent compound, 10m, displayed powerful anti-tumor potency in vitro, which may serve as a promising lead compound for further optimization.
The series of derivatives were prepared as shown in Scheme 1. The amine 8 was treated with acetic anhydride in pyridine to afford an intermediate, which was hydrogenated to give compound 9. Amide condensation of compound 9 with various acid afforded compounds 10a–w.
2. Results
2.1. Structured-Based Virtual Screening
Although imatinib or nilotinib displayed stronger synergic anti-proliferative effects with asciminib in vitro [17], a structural study has revealed that double-drugging with an “open” conformation binder resulted in an energetically frustrated ABL structure [20]. In order to discover new inhibitors with positive cooperativity with asciminib, a fully “closed” conformation of ABL (PDB id: 8SSN) was used. Commercial databases, including the SPECS database and ChemDiv database, approximately 2 million molecules, were docked into the kinase domain. The top-scored hits were combined and the common hits were selected. As drawn in Figure 2A, through a cascade of drug-likeness filtering, standard precision (SP) docking, extra precision (XP) docking, structural clustering, MD simulations and visual inspection, 11 structurally diverse compounds, namely A1*–*A11 (Figure 2B), were cherry-picked and purchased from the ChemDiv chemical library. These compounds were evaluated by parental Ba/F3 cells and Ba/F3 cells expressing BCR-ABL1 kinases, respectively. Initial inhibitory activity was conducted for each compound at concentrations of 50 μM (Figure 2C). Five out of eleven candidates showed inhibition against Ba/F3 (BCR-ABL1) cells (Figure S1). Among them, compound A8 showed inhibition potency at a lower concentration of 10 μM. We further measured the dose curves, and compound A8 displayed moderate antiproliferative activity with a GI_50_ of 6.4 μM (Figure 2D).
2.2. Similarity Search
Docking studies revealed that compound A8 formed crucial hydrogen bonds with Met-337 in the hinge region (Figure S2). To validate the predicted binding mode, a two-dimensional (2D) similarity search was conducted based on the benzothiazole moiety of A8 from the ChemDiv database, yielding compounds A12–A15. As shown in Table 1, A12 showed nearly two-fold decreased antiproliferative activity compared to A8 (10.7 μM vs. 6.4 μM), suggesting chlorine atom was preferred. The activity of compound A13 further decreased, suggesting the benzoyl group might clash with the hinge region. Compounds A14 and A15 showed a similar pattern. Compound A14 with methylsulfonyl displayed improved activity with a GI_50_ of 1.2 μM and was also cytotoxic. These findings suggested that the chemical structure of A8 might serve as a starting point for further exploration of novel BCR-ABL1 inhibitors.
2.3. Molecular Design
The docking study showed that compound A8 was embedded effectively well in the ATP-binding site, making polar contacts with Met-337 in the hinge region. The chlorobenzene moiety occupied a hydrophobic cavity (Figure 3A). To validate the predicted binding mode (Figure 3C,D), a 50-ns molecular dynamics (MD) simulation was performed. The simulation results demonstrated remarkable stability of compound A8 within the ATP-binding site, as evidenced by consistently low RMSD values. Notably, the benzo-thiazole scaffold maintained two characteristic hydrogen bonds with Met-337 throughout 99% of the simulation trajectory, further confirming the stability of the binding conformation. The ether oxygen of compound A8 formed a hydrogen bond with Asp-400, which was maintained for 53% of the simulation time. It was noticeable that a π-π interaction between the chlorobenzene moiety and Phe-401 was formed. We hypothesized that the strength of the π-π interaction is crucial for maintaining the protein’s “closed” conformation. Thus, the following two methods to study the structure–activity relationship (SAR) have been adopted: (1) small substituents were introduced to the benzene ring or (2) a conformation–restriction strategy was explored.
2.4. SAR Study
Guided by the predicted binding mode, we systematically explored the structure–activity relationship of this compound series (Table 2). First, we employed a conformation–restriction strategy, resulting in the loss of activity against Ba/F3 (BCR-ABL1) cells (compounds 10b–d). Derivative 10f, rather than 10e, which incorporated a naphthyl group, showed a slight increase in inhibitory activity. The decline of 10e may be attributed to increased polarity and steric hindrance. As shown in Figure S3A, compound 10f formed an edge-to-face π-π stacking interaction with Phe-401. These findings indicated that the angle and distance of the π-π interaction were important.
Next, small substituents were introduced into the benzene ring. The introduction of electron-withdrawing groups significantly enhanced BCR-ABL1 inhibitory activity (10g–m), whereas derivatives with an electron-donating methyl group showed a slight reduction in cellular potency (10s–w). Additionally, a directional “sigma-hole” formed by the chlorobenzene moiety in compound 10m led to increased binding affinity (Figures S3B and S4). Our analysis revealed that the presence of an oxygen atom was essential for inhibitory activity, as evidenced by the comparison between 10j–l and 10 n–p, which showed improved potency compared to 10a. Compounds 10n–p exhibited significantly reduced activity, likely due to the loss of the hydrogen bond with Asp-400. Notably, compounds 10q and 10r bearing the electron-withdrawing trifluoromethyl group were not very tolerant, which may be attributed to their relatively large volume. Building on these findings, dose-dependency curves of compounds 10f and 10m against multiple leukemia cell lines were determined. As shown in Table 3, compounds 10f and 10m showed an anti-tumor effect against Ba/F3 (BCR-ABL1) cells, with IC_50_ values of 2.2 and 0.63 μM, respectively. Compound 10m also showed limited activity against Ba/F3 (BCR-ABL^T315I^) cells.
With the aim of confirming the cooperative potency, K562 cells were treated with compound 10m and asciminib. As shown in Figure 4A, compounds 10m and 10f inhibited K562 cells with GI_50_ values of 0.98 and 2.7 μM, respectively. The synergistic effect of the drug combination was evaluated using the ZIP synergy score and calculated using SynergyFinder 3.0 [21]. As shown in Figure 4B,C, compound 10m displayed powerful synergistic activity with asciminib at 33.3 and 11.1 μM, with synergy score ranging from 10 to 58 (Table S3). However, when the concentration of imatinib was below 3.33 μM, the synergy scores were consistently below −10 (Table S2). Imatinib showed antagonistic effect in combination with asciminib, which was consistent with imatinib’s allosteric activator effect [22]. Furthermore, treatment with either compound 10m or asciminib alone significantly induced apoptosis in K562 cells, as quantitatively assessed through Annexin V/propidium iodide (PI) dual staining assays. Notably, the combination of 10m and asciminib demonstrated a synergistic effect, resulting in substantially enhanced apoptosis induction compared to either single agent treatment. These results strongly suggest the therapeutic potential of combination therapy utilizing both compounds.
To validate the inhibitory effects on BCR-ABL-mediated signaling pathways, we performed cellular assays using K562 cells treated with increasing concentrations of compound 10m or imatinib. The phosphorylation status of key downstream signaling molecules, including ABL1, STAT5, and CRKL was quantitatively analyzed through Western blot analysis (Figure 5 and Figure S3). A dose-dependent inhibition of p-ABL1, p-STAT5 and p-CRKL were observed. Compound 10m potently inhibited the BCR-ABL-dependent signaling pathway at 3.3 and 10 μM.
3. Discussion
The development of orthosteric BCR-ABL1 kinase inhibitors, such as imatinib, has significantly improved the prognosis of patients with CML or ALL [23,24]. The advancement of second- and third-generation BCR-ABL1 inhibitors has led to faster, deeper responses that predict superior outcomes [14,25]. With the breakthrough of the allosteric inhibitor asciminib [17], treatment is increasingly focused on identifying effective combination regimens. Although the simultaneous therapeutic blockade of orthosteric and allosteric inhibitors has shown anti-tumor activity against BCR-ABL1 point mutants and suppressed the emergence of resistant point mutations in vivo [18,19], structural studies have revealed that dual treatment with imatinib results in an energetically frustrated ABL structure. To address this, a fully inactive Abl structure [20] was applied to screen orthosteric BCR-ABL1 kinase inhibitors with synergistic effects in combination with asciminib. This approach led to the discovery of compound A8, which features a novel 2-acetamidobenzo[d]thiazol scaffold. Based on the A8 scaffold, 23 derivatives were synthesized and evaluated on Ba/F3 (BCR-ABL1) cells. The most potent compound, 10m, demonstrated anti-proliferative activity against Ba/F3 (BCR-ABL1) and K562 cells, with GI_50_ values of 0.63 and 0.98 μM, respectively. Compound 10m also exhibited potent inhibition of BCR-ABL signaling and showed synergistic effects on cell growth and apoptosis when combined with asciminib. These findings suggest that compound 10m may serve as a promising starting point for the development of dual-targeting therapeutic drugs for BCR-ABL1.
4. Experimental Section
4.1. Chemistry
General. All starting materials were obtained from commercial sources and used without additional purification. Unless otherwise specified, all reactions were performed without purification. Reaction progress was monitored by thin-layer chromatography (TLC) using silica gel plates (Yantai Jiangyou silica gel development Co. Ltd., HSGF254, Yantai, China), with visualization under UV light (254 nm or 365 nm) or by iodine vapor staining. Solvents were removed by rotary evaporation under reduced pressure at 40–45 °C. Nuclear magnetic resonance spectra (^1^H NMR and ^13^C NMR) were recorded on a Bruker 400 MHz spectrometer. Purification was carried out by flash column chromatography using silica gel (Qingdao Haiyang Chemical Co. Ltd., ZCX-II, 200–300 mesh, Qingdao, China) on a SepaBean^®^ machine U100 preparative liquid chromatography system. Compound purity was determined by HPLC analysis using an Agilent (Santa Clara, CA, USA) 1260 system (G7111A Quat pump and G7114A VWD detector) equipped with a Poroshell 120 EC-C18 reversed-phase column (4 μm) at 254 nM.
4.1.1. HPLC Method
A linear gradient program using water (solvent A) and acetonitrile (solvent B); t = 0–2 min, 10% B, t = 18 min, 90% B was employed. The flow rate was 1.2 mL/min and UV detection was set to 254 nm.
4.1.2. General Procedure for Synthesis of Compounds 10a–w
To a solution of acid (1.2 eq) in DMF (3 mL) was added BOP (1.5 eq) and DIPEA (3 eq). The reaction was stirred at 40 °C for 30 min, and then compound 9 (1.0 eq) was added. After the reaction was finished by TLC, the mixture was poured into water and extracted with ethyl acetate. The organic layer was dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified by silica gel column chromatography (DCM/MeOH, 30:1) followed by recrystallization in acetonitrile to provide the desired product.
4.1.3. N-(6-Aminobenzo[d]thiazol-2-yl)acetamide (9)
To a solution of 2-amino-6-nitrobenzothiazole (5.0 g, 25.61 mol) in pyridine (18 mL), acetic anhydride (15 mL, 135.17 mmol) was added. The reaction mixture was heated to 90 °C overnight. The reaction mixture was poured into 2N HCl (200 mL). The solid product that was formed was collected by filtration and washed with water and tert-butyl methyl ether and finally dried to give intermediate N-(6-nitrobenzo[d]thiazol-2-yl)acetamide as a pale yellow solid (5.1 g, yield 84%). ^1^H NMR (400 MHz, DMSO- d6 δ 12.76 (s, 1H), 9.02 (d, J = 2.4 Hz, 1H), 8.26 (dd, J = 8.8, 2.4 Hz, 1H), 7.87 (d, J = 8.8 Hz, 1H), 2.25 (s, 3H). ^13^C NMR (101 MHz, DMSO-d6) δ 170.64, 163.92, 153.92, 143.35, 132.60, 122.19, 120.98, 119.47, 23.30.
To a solution of compound N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (2.5 g, 10.54 mmol) in NH_4_OH (12 mL), a solution of sodium dithionite (8.8 g, 50.54 mmol) in water (40 mL) was quickly added; the reaction mixture was held at reflux overnight. After cooling, the crude product was filtered off, washed and dried under vacuum to afford compound 9 as a yellowish solid (1.7 g, yield 78%). ^1^H NMR (400 MHz, DMSO-d6) δ 11.88 (s, 1H), 7.39 (d, J = 8.8 Hz, 1H), 6.99 (s, 1H), 6.69 (d, J = 8.8 Hz, 1H), 5.14 (s, 2H), 2.14 (s, 3H). ^13^C NMR (101 MHz, DMSO-d6) δ 169.10, 153.53, 146.16, 140.07, 133.36, 121.27, 114.84, 104.55, 23.13.
4.1.4. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-phenoxyacetamide (10a)
Following general procedure A, compound 10a (82 mg, yield 76%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.22 (s, 1H), 8.31 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.61 (dd, J = 8.8, 2.0 Hz, 1H), 7.32 (t, J = 7.8 Hz, 2H), 7.08–6.93 (m, 3H), 4.72 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.74, 167.00, 158.29, 157.65, 145.37, 134.73, 132.32, 129.99, 121.67, 120.89, 119.61, 115.16, 112.76, 67.63, 23.19. R_T_ = 10.396 min, purity = 97.11%. Melting point: 156–158 °C.
4.1.5. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-phenoxypropanamide (10b)
Following general procedure A, compound 10b (88 mg, yield 72%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.28 (s, 1H), 10.26 (s, 1H), 8.29 (d, J = 2.0 Hz, 1H), 7.66 (d, J = 8.8 Hz, 1H), 7.59 (dd, J = 8.8, 2.0 Hz, 1H), 7.29 (tt, J = 7.6, 2.0 Hz, 2H), 7.02–6.91 (m, 3H), 4.95–4.85 (m, 1H), 2.19 (s, 3H), 1.57 (dd, J = 6.8, 2.0 Hz, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 170.49, 169.75, 157.75, 157.66, 145.34, 134.82, 132.29, 130.05, 121.65, 120.86, 119.55, 115.60, 112.69, 74.23, 23.19, 19.16. HRMS (ESI) [M + H]^+^ calcd for C18H18N3O3S: 356.10634; found: 356.10503. R_T_ = 10.722 min, purity = 97.47%. Melting point: 162–164 °C.
4.1.6. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2,3-dihydrobenzofuran-2-carboxamide (10c)
Following general procedure A, compound 10c (62 mg, yield 60%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.29 (s, 1H), 8.31 (d, J = 2.0 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.63 (dd, J = 8.8, 2.0 Hz, 1H), 7.25 (d, J = 7.6 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 6.92–6.81 (m, 2H), 5.34 (dd, J = 10.4, 6.8 Hz, 1H), 3.62–3.35 (m, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 169.48, 159.15, 157.72, 145.47, 134.66, 132.24, 128.41, 126.35, 125.45, 121.42, 120.84, 119.85, 113.05, 109.89, 80.84, 33.47, 23.19. HRMS (ESI) [M + H]^+^ calcd for C18H16N3O3S: 354.09069; found: 354.08949. R_T_ = 11.486 min, purity = 93.3%. Melting point: 163–165 °C.
4.1.7. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2,3-dihydrobenzo[b][1,4]dioxine-2-carboxamide (10d)
Following general procedure A, compound 10d (55 mg, yield 71%) was obtained as a yellowish solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.30 (s, 1H), 10.26 (s, 1H), 8.29 (s, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.60 (dd, J = 8.8, 2.0 Hz, 1H), 7.09–7.00 (m, 1H), 6.95–6.83 (m, 3H), 5.00 (dd, J = 6.0, 2.4 Hz, 1H), 4.54–4.32 (m, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.77, 165.98, 157.82, 145.56, 143.49, 142.87, 134.42, 132.28, 122.13, 121.99, 120.87, 119.81, 117.78, 117.51, 113.07, 73.14, 65.28, 23.19. HRMS (ESI) [M + H]^+^ calcd for C18H16N3O4S: 370.08560; found: 370.08429. R_T_ = 10.828 min, purity = 97.21%. Melting point: 166–168 °C.
4.1.8. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(naphthalen-1-yloxy)acetamide (10e)
Following general procedure A, compound 10e (83 mg, yield 75%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.31 (s, 1H), 10.37 (s, 1H), 8.43–8.29 (m, 2H), 7.88 (dd, J = 6.4, 3.6 Hz, 1H), 7.71 (d, J = 8.8 Hz, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.59–7.49 (m, 3H), 7.42 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 7.6 Hz, 1H), 4.96 (s, 2H), 2.21 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.76, 166.85, 157.72, 153.96, 145.43, 134.79, 134.58, 132.39, 127.88, 127.03, 126.52, 125.83, 125.39, 122.48, 121.15, 120.93, 119.68, 112.88, 106.08, 68.17, 23.20. HRMS (ESI) [M + H]^+^ calcd for C21H18N3O3S: 392.10634; found: 392.10484. R_T_ = 12.024 min, purity = 94.14%. Melting point: 174–175 °C.
4.1.9. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(naphthalen-2-yloxy)acetamide (10f)
Following general procedure A, compound 10f (66 mg, yield 68%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.29 (s, 1H), 8.33 (s, 1H), 7.92–7.78 (m, 3H), 7.70 (d, J = 8.8 Hz, 1H), 7.64 (d, J = 9.6 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.40–7.31 (m, 3H), 4.86 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.76, 166.85, 157.68, 156.19, 145.40, 134.74, 134.51, 132.33, 129.87, 129.25, 128.01, 127.24, 126.97, 124.36, 120.90, 119.69, 119.15, 112.85, 107.75, 67.77, 23.20. HRMS (ESI) [M + H]^+^ calcd for C21H18N3O3S: 392.10634; found: 392.10489. R_T_ = 11.883 min, purity = 99.89%. Melting point: 174–176 °C.
4.1.10. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(2-fluorophenoxy)acetamide (10g)
Following general procedure A, compound 10g (52 mg, yield 60%) was obtained as a yellowish solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.28 (s, 1H), 10.30 (s, 1H), 8.29 (s, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.58 (d, J = 8.8 Hz, 1H), 7.24 (dd, J = 12.0, 8.0 Hz, 1H), 7.19–7.07 (m, 2H), 7.05–6.94 (m, 1H), 4.82 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 168.14 (d, J = 320.7 Hz), 157.67, 153.41, 150.98, 146.39 (d, J = 10.3 Hz), 145.34, 134.74, 132.38, 125.21 (d, J = 3.6 Hz), 122.20 (d, J = 6.8 Hz), 120.94, 119.38, 116.67 (d, J = 17.8 Hz), 115.73, 112.56, 68.33, 23.18. ^19^F NMR (376 MHz, DMSO- d6) δ -134.19. HRMS (ESI) [M + H]^+^ calcd for C17H15FN3O3S: 360.08127; found: 360.07992. R_T_ = 10.465 min, purity = 98.42%. Melting point: 164–166 °C.
4.1.11. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(3-fluorophenoxy)acetamide (10h)
Following general procedure A, compound 10h (84 mg, yield 78%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.23 (s, 1H), 8.31 (s, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.34 (q, J = 8.0 Hz, 1H), 6.94–6.85 (m, 2H), 6.85–6.76 (m, 1H), 4.76 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) 168.15 (d, J = 318.6 Hz), 164.55, 162.14, 159.75 (d, J = 11.1 Hz), 157.71, 145.41, 134.65, 132.35, 131.19 (d, J =10.1 Hz), 120.89, 119.60, 112.79, 111.41 (d, J = 2.7 Hz), 108.31 (d, J = 21.0 Hz), 102.94 (d, J = 25.0 Hz). ^19^F NMR (376 MHz, DMSO- d6) δ -111.61. HRMS (ESI) [M + H]^+^ calcd for C17H15FN3O3S: 360.08127; found: 360.07997. R_T_ = 10.691 min, purity = 94.44%. Melting point: 163–165 °C.
4.1.12. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(4-fluorophenoxy)acetamide (10i)
Following general procedure A, compound 10i (82 mg, yield 80%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.21 (s, 1H), 8.29 (d, J = 2.0 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.60 (dd, J = 8.8, 2.0 Hz, 1H), 7.22–7.12 (m, 2H), 7.10–6.99 (m, 2H), 4.70 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 166.88, 156.15 (d, J = 302.8 Hz), 145.37, 134.69, 132.30, 120.89, 119.62, 116.61, 116.52, 116.46, 116.23, 112.78, 68.20, 23.19. ^19^F NMR (376 MHz, DMSO- d6) δ -123.25. HRMS (ESI) [M + H]^+^ calcd for C17H15FN3O3S: 360.08127; found: 360.07998. R_T_ = 10.342 min, purity = 99.41%. Melting point: 164–166 °C.
4.1.13. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(2-chlorophenoxy)acetamide (10j)
Following general procedure A, compound 10j (98 mg, yield 86%) was obtained as a yellowish solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.28 (s, 1H), 8.29 (d, J = 2.0 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.56 (dd, J = 8.8, 2.0 Hz, 1H), 7.46 (dd, J = 8.0, 1.6 Hz, 1H), 7.30 (td, J = 8.0, 1.6 Hz, 1H), 7.11 (d, J = 8.4 Hz, 1H), 6.99 (td, J = 8.0, 1.6 Hz, 1H), 4.86 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 166.35, 157.67, 153.93, 145.33, 134.73, 132.41, 130.56, 128.74, 122.59, 121.92, 120.99, 119.24, 114.55, 112.42, 68.16, 23.19. HRMS (ESI) [M + H]^+^ calcd for C17H15ClN3O3S: 376.05172; found: 376.05056. R_T_ = 10.768 min, purity = 99.98%. Melting point: 162–163 °C.
4.1.14. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(3-chlorophenoxy)acetamide (10k)
Following general procedure A, compound 10k (66 mg, yield 58%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.22 (s, 1H), 8.30 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.60 (dd, J = 8.8, 2.0 Hz, 1H), 7.33 (t, J = 8.2 Hz, 1H), 7.13 (t, J = 2.2 Hz, 1H), 7.02 (ddd, J = 10.9, 8.0, 2.2 Hz, 2H), 4.78 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.74, 166.55, 159.23, 157.71, 145.41, 134.64, 134.14, 132.35, 131.35, 121.63, 120.90, 119.60, 115.51, 114.12, 112.79, 67.73, 23.19. HRMS (ESI) [M + H]^+^ calcd for C17H15ClN3O3S: 376.05172; found: 376.05059. R_T_ = 11.407 min, purity = 97.07%. Melting point: 168–169 °C.
4.1.15. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(4-chlorophenoxy)acetamide (10l)
Following general procedure A, compound 10l (70 mg, yield 64%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.23 (s, 1H), 8.29 (d, J = 2.0 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.59 (dd, J = 8.8, 2.0 Hz, 1H), 7.44–7.32 (m, 2H), 7.12–6.98 (m, 2H), 4.74 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 166.67, 157.68, 157.18, 145.38, 134.67, 132.32, 129.73, 125.38, 120.90, 119.59, 116.98, 112.76, 67.82, 23.19. HRMS (ESI) [M + H]^+^ calcd for C17H15ClN3O3S: 376.05172; found: 376.05050. R_T_ = 11.327 min, purity = 98.07%. Melting point: 169–171 °C.
4.1.16. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(3,4-dichlorophenoxy)acetamide (10m)
Following general procedure A, compound 10m (72 mg, yield 68%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.23 (s, 1H), 8.28 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.62–7.54 (m, 2H), 7.34 (d, J = 2.8 Hz, 1H), 7.06 (dd, J = 8.8, 2.8 Hz, 1H), 4.79 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.76, 166.33, 157.80, 157.72, 145.42, 134.59, 132.34, 131.99, 131.45, 123.52, 120.92, 119.59, 117.41, 116.07, 112.79, 67.91, 23.20. HRMS (ESI) [M + H]^+^ calcd for C17H14Cl2N3O3S: 410.01274; found: 410.01160. R_T_ = 8.964 min, purity = 99.98%. Melting point: 172–174 °C.
4.1.17. N-(2-Acetamidobenzo[d]thiazol-6-yl)-3-(2-chlorophenyl)propanamide (10n)
Following general procedure A, compound 10n (52 mg, yield 60%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.26 (s, 1H), 10.09 (s, 1H), 8.29 (d, J = 2.0 Hz, 1H), 7.65 (d, J = 8.8 Hz, 1H), 7.50 (dd, J = 8.8, 2.0 Hz, 1H), 7.40 (ddd, J = 15.6, 7.6, 2.0 Hz, 2H), 7.32–7.20 (m, 2H), 3.04 (t, J = 7.6 Hz, 2H), 2.67 (t, J = 7.6 Hz, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 170.34, 169.68, 157.34, 144.90, 138.90, 135.60, 133.40, 132.36, 131.02, 129.70, 128.49, 127.75, 120.88, 118.97, 111.92, 36.34, 29.03, 23.18. R_T_ = 11.398 min, purity = 99.65%. Melting point: 165–166 °C.
4.1.18. N-(2-Acetamidobenzo[d]thiazol-6-yl)-3-(3-chlorophenyl)propanamide (10o)
Following general procedure A, compound 10o (90 mg, yield 74%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.27 (s, 1H), 10.05 (s, 1H), 8.26 (d, J = 2.0 Hz, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7.48 (dd, J = 8.8, 2.0 Hz, 1H), 7.36–7.20 (m, 4H), 2.94 (t, J = 7.6 Hz, 2H), 2.66 (t, J = 7.6 Hz, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 170.51, 169.69, 157.34, 144.89, 144.28, 135.59, 133.37, 132.36, 130.61, 128.67, 127.53, 126.43, 120.90, 118.94, 111.88, 37.94, 30.82, 23.19. HRMS (ESI) [M + H]^+^ calcd for C18H17ClN3O2S: 374.07245; found: 374.07135. R_T_ = 11.411 min, purity = 95.23%. Melting point: 162–164 °C.
4.1.19. N-(2-Acetamidobenzo[d]thiazol-6-yl)-3-(4-chlorophenyl)propanamide (10p)
Following general procedure A, compound 10p (71 mg, yield 63%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.26 (s, 1H), 10.05 (s, 1H), 8.27 (d, J = 2.0 Hz, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7.48 (dd, J = 8.8, 2.0 Hz, 1H), 7.38–7.24 (m, 4H), 2.92 (t, J = 7.6 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 170.55, 169.68, 157.33, 144.88, 140.68, 135.61, 132.35, 131.03, 130.64, 128.69, 120.89, 118.94, 111.87, 38.10, 30.55, 23.18. HRMS (ESI) [M + H]^+^ calcd for C18H17ClN3O2S: 374.07245; found: 374.07117. R_T_ = 11.329 min, purity = 99.1%. Melting point: 164–166 °C.
4.1.20. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(3-(trifluoromethyl)phenoxy) acetamide (10q)
Following general procedure A, compound 10q (42 mg, yield 36%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.30 (s, 1H), 10.26 (s, 1H), 8.30 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.61 (dd, J = 8.8, 2.0 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.40–7.29 (m, 3H), 4.85 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 166.50, 158.58, 157.72, 145.44, 134.61, 132.35, 131.20, 130.72 (d, J = 31.6 Hz), 124.44 (d, J = 270.7 Hz), 120.90, 119.63, 119.20, 118.15 (q, J = 3.8 Hz), 112.84, 112.19 (q, J = 3.8 Hz), 67.71, 23.17. ^19^F NMR (376 MHz, DMSO- d6) δ -61.15. HRMS (ESI) [M + H]^+^ calcd for C18H15F3N3O3S: 410.07807; found: 410.07646. R_T_ = 10.481 min, purity = 98.96%. Melting point: 170–172 °C.
4.1.21. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(4-(trifluoromethyl)phenoxy) acetamide (10r)
Following general procedure A, compound 10r (35 mg, yield 33%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.30 (s, 1H), 8.29 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 3H), 7.59 (dd, J = 8.8, 2.0 Hz, 1H), 7.20 (d, J = 8.8 Hz, 2H), 4.85 (s, 2H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 166.39, 161.19, 157.70, 145.39, 134.65, 132.34, 127.43 (q, J = 360.0 Hz), 124.98 (d, J = 269.4 Hz), 122.16 (d, J = 31.9 Hz), 120.92, 119.55, 115.70, 112.75, 67.54, 23.19. ^19^F NMR (376 MHz, DMSO- d6) δ -59.86. HRMS (ESI) [M + H]^+^ calcd for C18H15F3N3O3S: 410.07807; found: 410.07662. R_T_ = 11.814 min, purity = 96.70%. Melting point: 168–170 °C.
4.1.22. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(o-tolyloxy)acetamide (10s)
Following general procedure A, compound 10s (73 mg, yield 55%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.18 (s, 1H), 8.31 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.59 (dd, J = 8.8, 2.0 Hz, 1H), 7.21–7.11 (m, 2H), 6.93–6.83 (m, 2H), 4.74 (s, 2H), 2.26 (s, 3H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.74, 167.12, 157.66, 156.53, 145.34, 134.78, 132.37, 131.09, 127.38, 126.65, 121.38, 120.92, 119.48, 112.63, 111.97, 67.94, 23.19, 16.63. HRMS (ESI) [M + H]^+^ calcd for C18H18N3O3S: 356.10634; found: 356.10494. R_T_ = 11.336 min, purity = 98.76%. Melting point: 164–165 °C.
4.1.23. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(m-tolyloxy)acetamide (10t)
Following general procedure A, compound 10t (59 mg, yield 60%) was obtained as a yellowish solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.30 (s, 1H), 10.20 (s, 1H), 8.32 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.63 (dd, J = 8.8, 2.0 Hz, 1H), 7.19 (t, J = 8.0 Hz, 1H), 6.89–6.76 (m, 3H), 4.70 (s, 2H), 2.29 (s, 3H), 2.20 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 167.06, 158.31, 157.66, 145.37, 139.48, 134.74, 132.32, 129.71, 122.44, 120.89, 119.62, 115.93, 112.77, 112.06, 67.62, 23.19, 21.58. HRMS (ESI) [M + H]^+^ calcd for C18H18N3O3S: 356.10634; found: 356.10510. R_T_ = 9.658 min, purity = 99.98%. Melting point: 166–168 °C.
4.1.24. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(p-tolyloxy)acetamide (10u)
Following general procedure A, compound 10u (62 mg, yield 48%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.19 (s, 1H), 8.29 (d, J = 2.4 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.60 (dd, J = 8.8, 2.4 Hz, 1H), 7.11 (d, J = 8.4 Hz, 2H), 6.96–6.84 (m, 2H), 4.67 (s, 2H), 2.23 (s, 3H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 167.13, 157.64, 156.21, 145.34, 134.74, 132.29, 130.41, 130.31, 120.88, 119.61, 115.03, 112.74, 67.83, 23.20, 20.55. HRMS (ESI) [M + H]^+^ calcd for C18H18N3O3S: 356.10634; found: 356.10507. R_T_ = 11.162 min, purity = 99.98%. Melting point: 163–164 °C.
4.1.25. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(3-methoxyphenoxy)acetamide (10v)
Following general procedure A, compound 10v (62 mg, yield 48%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.21 (s, 1H), 8.30 (d, J = 2.0 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.61 (dd, J = 8.8, 2.0 Hz, 1H), 7.25–7.17 (m, 1H), 6.68–6.49 (m, 3H), 4.70 (s, 2H), 3.74 (s, 3H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 166.92, 160.92, 159.48, 157.67, 145.37, 134.70, 132.31, 130.49, 120.89, 119.62, 112.78, 107.36, 107.24, 101.63, 67.73, 55.60, 23.19. HRMS (ESI) [M + H]^+^ calcd for C18H18N3O4S: 372.10125; found: 372.09975. R_T_ = 11.370 min, purity = 99.98%. Melting point: 165–167 °C.
4.1.26. N-(2-Acetamidobenzo[d]thiazol-6-yl)-2-(4-methoxyphenoxy)acetamide (10w)
Following general procedure A, compound 10w (90 mg, yield 76%) was obtained as a white solid. ^1^H NMR (400 MHz, DMSO- d6) δ 12.29 (s, 1H), 10.17 (s, 1H), 8.31 (d, J = 2.0 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.62 (dd, J = 8.8, 2.0 Hz, 1H), 6.97 (d, J = 9.2 Hz, 2H), 6.88 (d, J = 9.2 Hz, 2H), 4.64 (s, 2H), 3.69 (s, 3H), 2.19 (s, 3H). ^13^C NMR (101 MHz, DMSO- d6) δ 169.75, 167.24, 157.64, 154.32, 152.28, 145.35, 134.73, 132.29, 120.87, 119.65, 116.20, 115.07, 112.77, 68.42, 55.82, 23.19. HRMS (ESI) [M + H]^+^ calcd for C18H18N3O4S: 372.10125; found: 372.09988. R_T_ = 10.215 min, purity = 94.20%. Melting point: 165–167 °C.
4.2. Viability Assay
Cell viability was assessed using the Cell Counting Kit-8 (CCK-8) assay. Cells were seeded in 96-well plates at a density of 10,000 cells per well in RPMI 1640 medium. Test compounds were prepared by serially diluting stock solutions 3-fold in DMSO to generate an eight-point concentration gradient (100 μM to 0.045 μM), followed by addition to the cultured cells. After 72 h of incubation, 10 μL of CCK-8 reagent was added to each well, and the plates were further incubated for 2–4 h. Absorbance was measured at 450 nm using a microplate reader. Dose-response curves were generated and analyzed using GraphPad Prism 8 (version 8.4.3.686).
4.3. Apoptosis Assays
Cell apoptosis was evaluated through Annexin V and propidium iodide (PI) dual staining using an Annexin V-FITC Apoptosis Detection Kit (Vazyme, #A211-02, Nanjing, China) following the manufacturer’s protocol. Cells were seeded at a density of 500,000 cells per well in 12-well plates containing RPMI 1640 medium. K562 cells were treated with either compound 10m or asciminib for 48 or 72 h. Apoptotic signals were quantified using a FACS Calibur flow cytometer (Cy-toFLEX).
4.4. Western Blot Analysis
Western blot analysis was conducted following established protocols. K562 cells were treated with varying concentrations of the test compound for 60 min, followed by protein extraction using RIPA buffer. Protein quantification was performed using bicinchoninic acid assay, and 30–60 μg of protein samples were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis before being transferred onto nitrocellulose membranes. Membranes were probed with primary antibodies overnight at 4 °C. Following incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (Thermo Scientific, Waltham, MA, USA), protein bands were detected using a chemiluminescent HRP substrate (Millipore Corporation, #WBKLS0500, Billerica, MA, USA) and imaged with a Syngene GeneGnome-XRQ-NPC imager (Synoptics, Britain). Quantitative analysis was performed by measuring band intensity using ImageJ (version 1.43) software. The following primary antibodies were used: c-Abl (ABclonal, #A0282, Woburn, MA, USA), phosphor-c-Abl (ABclonal, #AP0001), STAT5 (ABclonal, #A5029), phosphor-STAT5 (ABclonal, #AP0887), CRKL (ABclonal, #A11735), and phospho-CRKL (ABclonal, #AP0824).
4.5. Molecular Docking and MD Simulations
The protein structure was retrieved from the Protein Data Bank (http://www.rcsb.org/pdb, accessed on 10 December 2024). The X-ray crystal structure of BCR-ABL (PDB id: 8SSN) was selected for molecular docking studies using Maestro (Schrödinger, LLC, New York, NY, USA, 2019). Ligand docking was performed in the orthosteric binding pocket using Glide. Based on the docking results, separate 50 ns molecular dynamics simulations were conducted with Desmond. To maintain system neutrality, Na^+^ and Cl^−^ ions were added at physiological concentration (0.15 mol·L^−1^). Simulations utilized the OPLS3 force field and TIP3P explicit solvent model, resulting in a solvated system containing approximately ~37,000 atoms. Data were recorded at 20 ps intervals under NPT ensemble conditions, with temperature maintained at 300 K and pressure at 1.01 bar. Ligand-protein interactions were analyzed using the Simulation Interactions Diagram tool. For visual inspection, MD simulations were employed as the final step in the virtual screening pipeline. After the ChemDiv and Specs commercial compound libraries were processed through Glide SP and XP docking, a 20 ns MD simulation was used to filter and identify the final candidate compounds, with the evaluation criteria including: stable RMSD values of the heavy atoms of the small molecules relative to the backbone atoms of the kinase throughout the simulation (<2 Å), sustained hydrogen bond interactions with Met-337, and prolonged π-π interactions with Phe-401.
4.6. Synergy Study
SynergyFinder 3.0 (https://synergyfinder.fimm.fi, accessed on 10 December 2024) is a web application for interactive analysis and visualization of multi-drug combination response data. Cells were plated at a density of 10,000 cells per well in 96-well plates containing RPMI 1640 medium. Imatinib or compound 10m was administered in combination with asciminib, with the concentrations of test compounds added as specified in Figure 4B,C. Following a 72 h incubation period, 10 μL of CCK-8 dye was added to each well, and the cells were further incubated for 2–4 h. Fluorescence intensity was subsequently measured at 450 nm. The drug combination effects were quantified as percentage inhibition of cell viability. The experimental data were analyzed using SynergyFinder 3.0, with a four-parameter logistic regression (LL4) model selected for curve fitting and data analysis. The Synergy Score based on the ZIP (Zero Interaction Potency) model is used to evaluate the synergistic effects of drug combinations.
4.7. Reagents and Compounds
Compounds used for BCR-ABL1 inhibitors screening (A1–A15, see Table S1) were purchased from the ChemDiv (https://www.chemdiv.com, accessed on 10 December 2024) with a purity of more than 95%. Imatinib, ponatinib, and asciminib were purchased from Bidepharm Co., Ltd. (Shanghai, China).
5. Conclusions
In conclusion, the combination of orthosteric and allosteric BCR-ABL1 inhibitors holds great promise for overcoming resistance in CML and ALL treatment. Structure-based virtual screening and SAR studies led to the identification of compound A8, featuring a novel 2-acetamidobenzo[d]thiazol scaffold. Derivatives of A8, particularly compound 10m, demonstrated potent anti-proliferative activity, inhibited BCR-ABL signaling, and exhibited synergistic effects with asciminib in cell growth and apoptosis. These findings highlight compound 10m as a potential lead for developing dual-targeting BCR-ABL1 therapeutic agents.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia Nat. Rev. Cancer 2005517218310.1038/nrc 156715719031 · doi ↗ · pubmed ↗
- 2Soverini S. Bassan R. Lion T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: Recent advances and remaining challenges J. Hematol. Oncol.2019123910.1186/s 13045-019-0729-231014376 PMC 6480772 · doi ↗ · pubmed ↗
- 3Dong Y. Shi O. Zeng Q. Lu X. Wang W. Li Y. Wang Q. Leukemia incidence trends at the global, regional, and national level between 1990 and 2017 Exp. Hematol. Oncol.202091410.1186/s 40164-020-00170-632577323 PMC 7304189 · doi ↗ · pubmed ↗
- 4Quintás-Cardama A. Cortes J. Molecular biology of bcr-abl 1-positive chronic myeloid leukemia Blood 20091131619163010.1182/blood-2008-03-14479018827185 PMC 3952549 · doi ↗ · pubmed ↗
- 5Heinrich M.C. Griffith D.J. Druker B.J. Wait C.L. Ott K.A. Zigler A.J. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor Blood 20009692593210.1182/blood.V 96.3.92510910906 · doi ↗ · pubmed ↗
- 6Druker B.J. Sawyers C.L. Kantarjian H. Resta D.J. Reese S.F. Ford J.M. Capdeville R. Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome N. Engl. J. Med.20013441038104210.1056/NEJM 20010405344140211287973 · doi ↗ · pubmed ↗
- 7Weisberg E. Manley P.W. Breitenstein W. Brüggen J. Cowan-Jacob S.W. Ray A. Huntly B. Fabbro D. Fendrich G. Hall-Meyers E. Characterization of AMN 107, a selective inhibitor of native and mutant Bcr-Abl Cancer Cell 2005712914110.1016/j.ccr.2005.01.00715710326 · doi ↗ · pubmed ↗
- 8Copland M. Hamilton A. Elrick L.J. Baird J.W. Allan E.K. Jordanides N. Barow M. Mountford J.C. Holyoake T.L. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction Blood 20061074532453910.1182/blood-2005-07-294716469872 · doi ↗ · pubmed ↗