Computational and Experimental Studies on the α-Functionalization of Ketones Using Domino Reactions: A Strategy to Increase Chemoselectivity at the α-Carbon of Ketones
Hui Sun, Li-Heng Yang, Meng-Yun Fu, Bin Cui

TL;DR
This paper introduces a new method to improve the selectivity of chemical reactions involving ketones using TMSCF3, enhancing efficiency and reducing by-products.
Contribution
The study proposes a novel strategy to increase chemoselectivity in domino reactions through intermolecular interactions and TMSCF3.
Findings
TMSCF3 increases the activation energy difference between main and side reactions, improving chemoselectivity.
The recovery rate of TMSCF3 through rectification was 87.2%, indicating its reusability.
DFT calculations and experiments confirmed the mechanism's validity.
Abstract
A facile strategy to increase the chemoselectivity of domino reactions was proposed and successfully applied in the α-functionalization of ketones. The strategy involved widening the activation energy of the main reaction and side reaction through intermolecular interactions, thereby increasing the chemoselectivity of the domino reaction. In the proposed α-functionalization reaction, TMSCF3 acted as an excellent reagent which increased the nucleophilicity of DMF through the Van der Waals force and reduced the nucleophilicity of H2O through a hydrogen bond. We found that TMSCF3 can increase the activation energy difference between the main reaction and side reaction using DFT calculations, which greatly increased chemoselectivity and avoided the formation of by-products. TMSCF3 was recycled by rectification, and the average recovery rate was 87.2%. DFT calculations, XRD experiments, and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
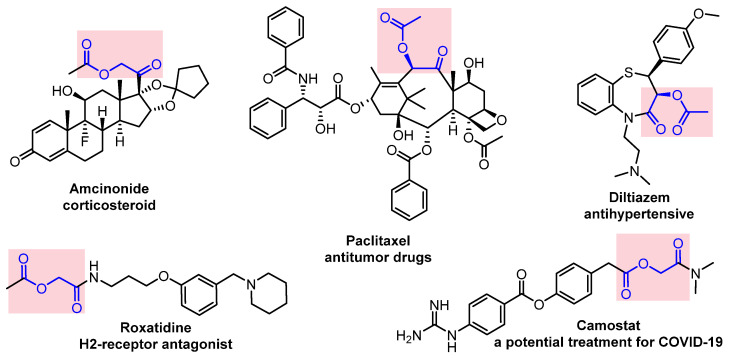 Figure 1
Figure 1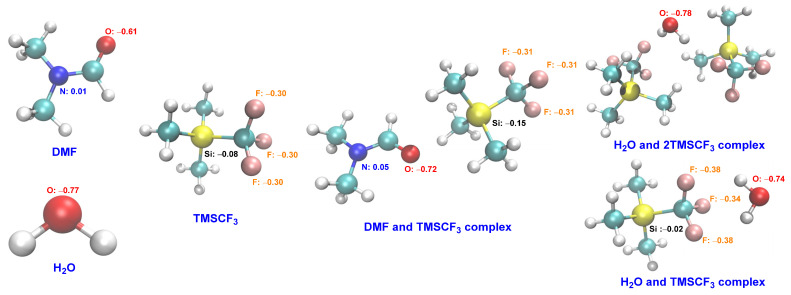 Figure 2
Figure 2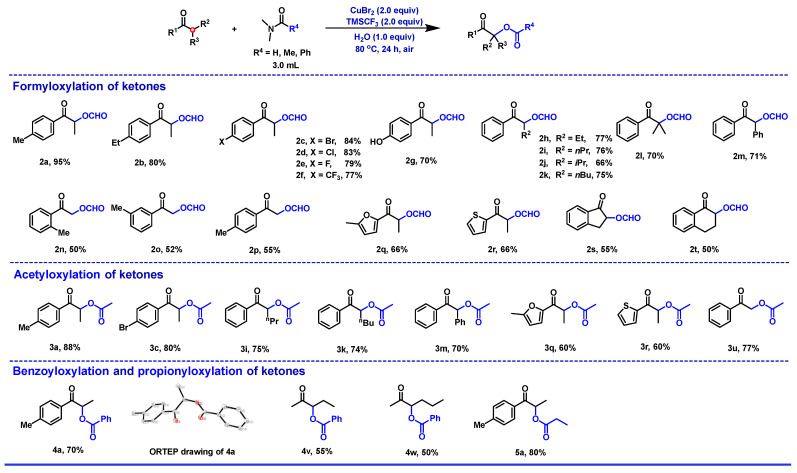 Figure 3
Figure 3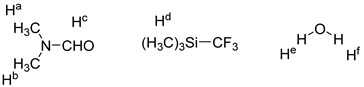 Figure 4
Figure 4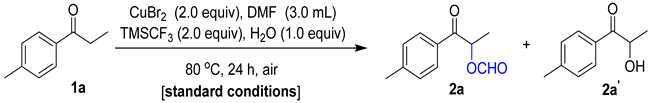 Figure 5
Figure 5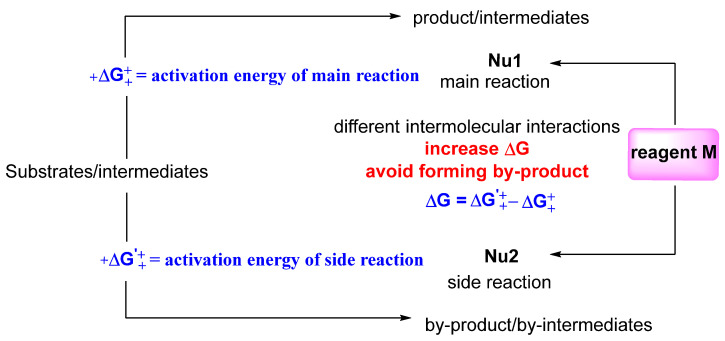 Figure 6
Figure 6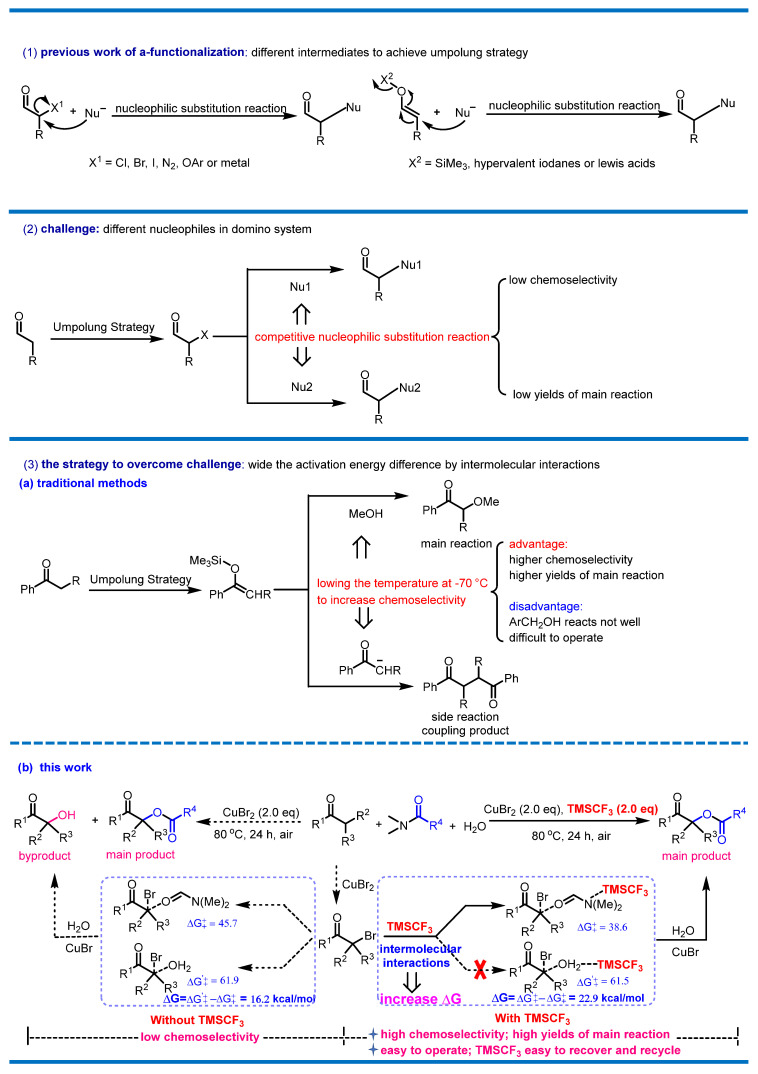 Figure 7
Figure 7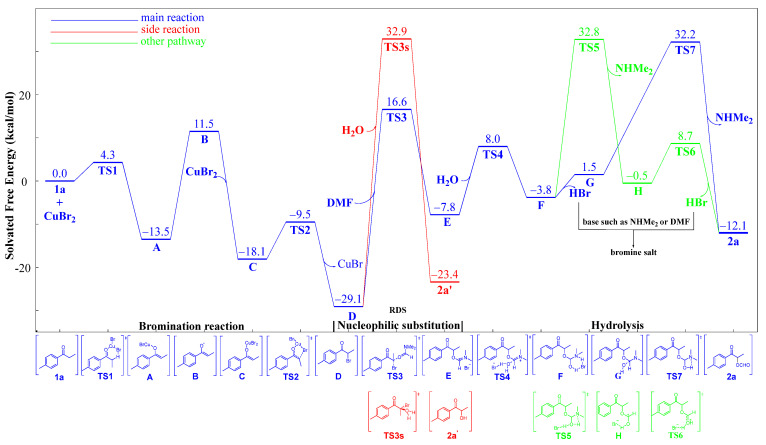 Figure 8
Figure 8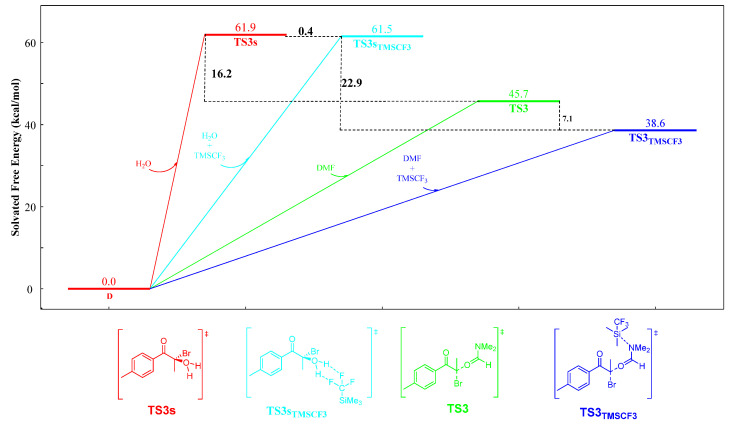 Figure 9
Figure 9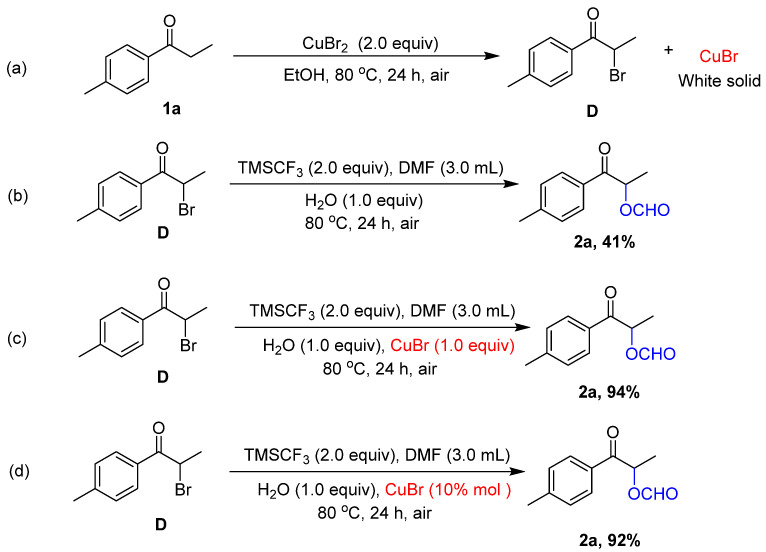 Figure 10
Figure 10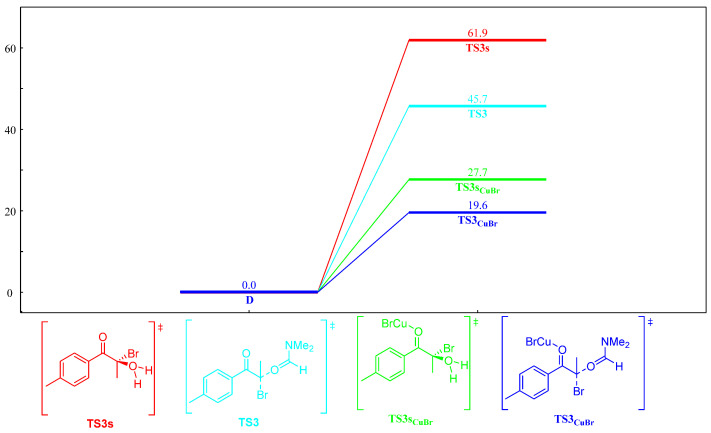 Figure 11
Figure 11- —National Natural Science Foundation of China
- —Natural Science Foundation of Hebei Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChemical Synthesis and Analysis · Cyclopropane Reaction Mechanisms · Asymmetric Synthesis and Catalysis
1. Introduction
Domino reactions incorporate two or more consecutive transformations in a single reaction vessel, in which the formed functionalities in the previous reaction steps are used in subsequent transformations without the addition of other materials [1,2,3,4,5]. Given that domino reactions proceed without the isolation of any intermediates, they have the advantages of atom-economy, reduced generation of waste, and the saving of materials, energy and time. Nevertheless, domino reactions often bring about side reactions [2,3,4,5,6,7]. The main reason for this is that domino reactions cannot guarantee that the product of the previous step will only be the next step without the inclusion of other steps within such complex reaction conditions. In order to solve the chemoselectivity problem of domino reactions, we conceived a facile strategy, as shown in Scheme 1. We added reagent M into the domino reaction system to expand the activation energy of the main reaction and side reaction through intermolecular interactions, thereby increasing chemoselectivity and yields (Scheme 1). Under the guidance of the proposed strategy, we carried out a series of explorations to prove the feasibility of the strategy. As a result, we successfully designed an α-functionalization method involving a carbonyl compound reaction via the bromination/nucleophilic substitution/hydrolysis sequence based on the above-mentioned strategy. We believe that this strategy has the potential to fundamentally reshape the landscape of multi-step synthetic operations, with the ability to convert reactions previously limited by low chemoselectivity into highly chemoselective domino transformations. Beyond simply enhancing chemoselectivity and improving main product yields, this strategy provides both a theoretical framework and practical case studies for conceiving and developing domino synthetic methodologies. In doing so, it not only delivers tangible advancements in application but also encourages broader innovation in domino synthesis.
Many α-functionalization reactions of carbonyl compounds, which have been the subject of increasing interest in recent decades, are cascade reactions that require the reversal of the polarity of carbonyl compounds [8,9]. In conversion, functionalization at the α-position can be achieved with the help of various intermediates [10,11,12,13], such as halogens [14,15,16,17,18,19,20,21,22,23,24], hypervalent iodine [25,26,27,28,29], silyl enol ethers, ethers [30], α-diazo ketones [31,32,33,34], Lewis acids [35], and transition-metal catalysts (Scheme 2, (1)) [36,37]. These intermediates obtain the target compounds through the nucleophilic substitution process. Due to the presence of multiple nucleophiles in the domino reaction system, the nucleophilic substitution process may produce undesirable by-products, resulting in low chemoselectivity and yields (Scheme 2, (2)). In previous studies, many reactions needed to be achieved through the multi-step reaction process instead of the domino strategy in order to increase chemoselectivity [38,39,40,41,42,43]. Although many reactions were tolerated through the domino strategy, these studies required extreme measures to increase chemoselectivity, such as lowering the temperature [44,45], which can reduce reactivity and the reaction scope (Scheme 2, (3a)). Compared with these traditional strategies used to increase chemoselectivity, our proposed strategy does not sacrifice excellent reactivity and the reaction scope in exchange for relatively high chemoselectivity (Scheme 2, (3a)). Therefore, we applied the proposed strategy to the α-functionalization of ketones in order to increase chemoselectivity without affecting its reactivity and the reaction scope (Scheme 2, (3b)).
As seen in the above-mentioned works regarding the α-functionalization of ketones, α-acyloxycarbonyl compounds are versatile synthons for the synthesis of various cyclic and heterocyclic natural products and pharmaceuticals [46]. A number of classic drugs, lead compounds, anti-inflammatory drugs [47], antitumor drugs, antihypertensive drugs, H2 receptor antagonists, and potential treatment drugs for COVID-19 [48,49,50] contain α-acyloxycarbonyl group (Figure 1). The development of a green and practical methodology using the domino strategy is needed for the preparation of α-acyloxy ketones.
2. Results and Discussion
In order to increase the chemoselectivity of the reaction and reduce the yield of by-products, the mechanism of the acyloxylation domino reaction was studied by DFT calculations (Scheme 3). Computational and experimental studies showed that nucleophilic substitution is the main process for the production of by-product 2a′ (Scheme 3, nucleophilic substitution). The reason for this is that the domino system contained both H_2_O and DMF nucleophiles, and the Gibbs free energy difference of TS3 and TS3s was 16.3 kcal/mol, which caused the side reaction to proceed under 80 °C. In order to improve the chemoselectivity of the reaction, we explored a suitable method to reduce the Gibbs free energy of TS3 or to increase the Gibbs free energy of TS3s, thereby making the barrier larger between the Gibbs free energy of TS3 and TS3s.
In our previous work, we found that DMF and TMSCF_3_ reagents interact together and to successfully activate trifluoromethyl [51,52]. The ^1^H NMR experiments were carried out to study the possible interaction between H_2_O and TMSCF_3_. Upfield shifts were observed for protons of H_2_O when H_2_O and TMSCF_3_ were mixed in CDCl_3_ (Table 1). Our previous work and ^1^H NMR experiments indicated that an interaction existed between TMSCF_3_, H_2_O, and DMF. When using the DFT calculation for the DMF and TMSCF_3_ complex, we found that the oxygen’s charge [53,54,55] of the DMF and TMSCF_3_ complex was increased compared with that of DMF (Figure 2, the DMF and TMSCF_3_ complex vs. DMF). The oxygen’s charge [53,54,55] of the H_2_O and TMSCF_3_ complex, which was formed through hydrogen bonding, was slightly reduced compared with that of H_2_O (Figure 2, the H_2_O and TMSCF_3_ complex vs. H_2_O). Similarly, the condensed Fukui functions and RDG proved that the results are consistent with the charge analysis method (the details of this are in the Supplementary Materials: Part 2. the condensed Fukui functions and RDG of DMF, H_2_O, and their complexes with TMSCF_3_). These results laterally showed that the addition of TMSCF_3_ increases the nucleophilicity of DMF and reduces the nucleophilicity of H_2_O.
In order to further prove the rationality of the strategy, we calculated the nucleophilic substitution of the domino reaction under TMSCF_3_ conditions, and the results are shown in Scheme 4. The results show that the activation energy decreased after adding TMSCF_3_ (Scheme 4, TS3_TMSCF3_ vs. TS3 and TS3s_TMSCF3_ vs. TS3s), and the Gibbs free energy increased to 22.9 kcal/mol (Scheme 4, TS3_TMSCF3_ vs. TS3s_TMSCF3_). In the process of the nucleophilic substitution reaction, DMF had a greater advantage and it was difficult or impossible to react H_2_O because of its excessively high activation energy (Scheme 4, TS3_TMSCF3_ vs. TS3s_TMSCF3_). Therefore, this strategy can prevent the formation of by-products and can increase chemoselectivity. The enhancement of the chemoselectivity of the nucleophilic substitution process was beneficial for the subsequent hydrolysis process, which required the participation of H_2_O (Scheme 3, hydrolysis).
On the basis of the DFT calculation results, we surveyed a variety of reaction conditions using 1a and DMF as a model system, as seen in Table 2. We found that CuBr_2_ effectively participates in the required reaction of 1a and DMF to provide 2a under TMSCF_3_ conditions, and the by-product 2a′ was not detected (Table 2, entry 1). An amount of 65% of the yield of 2a and 23% of the yield of by-product 2a′ was obtained without TMSCF_3,_ which is consistent with the result of the DFT calculations (Table 2, entry 2). The results were unsatisfactory when other trimethylsilyl derivatives were used (Table S1 in Supplementary Materials). Other halogenated copper salts were used instead of CuBr_2,_ and the result was not satisfactory (Table 2, entries 3–6). Reactions without CuBr_2_ were not successful (Table 2, entry 7). A further reduction in the amount of CuBr_2_ from 2.0 equiv to 1.0 equiv led to a drop in substrate conversions (Table 2, entries 1 vs. 8) and the increase in the amount of CuBr_2_ from 2.0 equiv to 3.0 equiv had little effect on the substrate conversions (Table 2, entries 1 vs. 9). The reaction was suppressed without H_2_O (Table 2, entry 10) and the reaction had little effect on the Ar protection condition compared with the air condition (Table 2, entry 11). Therefore, the α-formyloxylation reaction was finally carried out in the standard conditions.
Having identified the optimal reaction conditions, various substituted ketones were evaluated to determine their influence on the overall reactivity and to establish the reaction scope (Figure 3). In general, the α-formyloxylation methodology was amenable to a range of aryl-substituted α-formyloxy ketones that bear various electron-withdrawing and electron-donating substituents, furnishing the corresponding products in typically good-to-excellent yields (Figure 3, 2a–2g). The substrates of ortho-methyl (1n), meta-methyl (1o), and para-methyl (1p) substituents on the benzene rings did not significantly influence the reaction process. The steric effect of aromatic and aliphatic ketones was investigated and the results indicate that the small groups were slightly superior to the bulky groups in the reactions (Figure 3, 2h–2m). Other aromatic rings of the aromatic ketones, such as the furan ring and the thiophene ring, had little effect on the course of the reaction, and the resulting products were obtained in good isolated yields (Figure 3, 2q–2r). Cyclic ketones such as 1s and 1t obtained corresponding products in moderate yields. To prove the scalability of the reaction, substrate 1a was carried out on a gram scale, and the desired product 2a was obtained in a 63% isolated yield. By slowly adding CuBr_2_ without changing the amounts of other materials, the yield increased to 92%. After the α-formyloxylation of the ketones, the α-acyloxylation of the ketones was included within the reaction with N,N-dimethylamides (R^4^ = Me, Ph, and Et) instead of DMF (Figure 3, 3, 4 and 5). N,N-dimethylamides (R^4^ = Me, Ph, and Et) displayed good reactivity under standard conditions, and the results indicated that the size of the acyl group on the N,N-dimethylamides had little effect on the course of the reaction (Figure 3, 2, 3, 4, and 5). Compound 4a was subjected to an X-ray diffraction experiment to determine the skeleton, and the ORTEP drawing clearly confirmed the structure (Figure S2 in the Supplementary Materials).
Although DFT calculations had theoretically provided the domino reaction mechanism (Scheme 3), it was necessary to further verify the domino reaction process through experiments. The designed domino reaction had three reaction processes, namely bromination, nucleophilic substitution, and hydrolysis. We adopted a segmented method to verify the reaction process in Scheme 5. To ensure that the reaction first took place through the bromination reaction process, we added 1a, CuBr_2,_ without any other substances to the reaction system. The brominated product D and a white solid powder were obtained after careful separation and purification (Scheme 5, (a)). Based on the designed reaction mechanism and the results of DFT calculations, we guessed that the white solid powder was CuBr. Further XRD studies of the white solid powder indicated that it was CuBr (Figure S1 in the Supplementary Materials). It was proven that the bromination reaction process was tolerated because of the designed mechanism. Then, we added D, DMF, TMSCF_3,_ and H_2_O to the reaction system, which performed nucleophilic substitution and hydrolysis processes. We found that the reaction was achieved, but the yield of 2a was significantly lower than that in the domino reaction system (Scheme 5, (b)). We guessed that the product CuBr of the previous bromination process promoted the subsequent processes. CuBr was added to the reaction system of (b), and we found that it greatly promoted the processes of nucleophilic substitution and hydrolysis (Scheme 5, (b) vs. (c)). It was noteworthy that CuBr displayed good reactivity, and 10% CuBr was shown to be successful (Scheme 5, (d)).
To obtain detailed insight into the role of CuBr in the domino reaction, control experiments and DFT calculations were carried out, as seen in Scheme 6. CuBr greatly reduced the activation energy of the nucleophilic substitution process (Scheme 6, TS3_CuBr_ vs. TS3 and TS3s_CuBr_ vs. TS3s). However, CuBr had little effect on the activation energy difference between the main reaction and the side reaction (Scheme 6, the difference between TS3 and TS3s vs. the difference between TS3_CuBr_ and TS3s_CuBr_), which did not increase chemoselectivity.
TMSCF_3_ played a key role in increasing the chemoselectivity of the reaction. We successfully recovered TMSCF_3_ in the reaction system through rectification (Figure S3 in the Supplementary Materials). This showed that TMSCF_3_ could be recovered and reused, which embodies the concept of atomic economy.
3. Conclusions
In summary, a facile strategy was proposed to increase the chemoselectivity of domino reactions, and this strategy was used to design an α-formyloxylation and an α-acyloxylation domino reaction of ketones. The α-acyloxylation of ketones proved the feasibility of the strategy. TMSCF_3,_ when added to the reaction system, enlarged the activation energy difference between the main reaction and side reaction through intermolecular interactions, thereby increasing chemoselectivity and avoiding the formation of by-products. TMSCF_3_ was recovered and reused, which embodies the concept of atomic economy. The chemoselectivity problem of other multi-step reactions or domino reactions solved by this strategy is under further study.
4. Materials and Methods
4.1. General Experimental Information
All reactions were carried out in sealed tube at 80 °C under air, unless stated otherwise. The purchased reagents were of the highest commercial quality and were used without further purification, unless otherwise stated. DMF and DMA were dried by CaCl_2_ and vigorously stirred with CaH_2_ overnight; then, this mixture was distilled out and sealed using 4 Å molecular sieves for storage under N_2_ protection. The reagents were weighed using a Sartorius analytical balance (BCE224-1CCN) with a precision of 0.0001 g. The ^1^H and ^13^C NMR spectra were recorded on a Bruker DRX 500 (or 400) spectrometer at 298 K using deuterated chloroform as the solvent and TMS as the internal reference. The ^1^H NMR were recorded as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, coupling constant (s) in Hz, integration). The data for ^13^C NMR are reported in terms of chemical shift (δ, ppm). The reactions were monitored through thin layer chromatography (TLC) on silica gel plates (GF 254) using a mixture of iodine and silica gel as the visualizing agent, unless otherwise stated. Flash column chromatography was performed using silica gel (200–400 meshes). The infrared (IR) spectra were recorded with a KBr pellet, and the wavenumbers were given in cm^−1^. HRMS analyses were carried out with Varian FTICR-MS 7.0 T. The melting points were obtained on a Mettler Toledo MP50 apparatus.
4.2. General Procedure for Acyloxylation Reaction
Ketone (1.0 mmol), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to a sealed tube (40 mL). Amide (3.0 mL) was added, and the mixture was stirred vigorously at 80 °C for 24 h. After completion, the reaction mixture was added into DCM (30 mL) and washed with NaCl solution (50 mL × 3). The organic mixture was dried with MgSO_4_, concentrated in vacuo, and separated with flash column chromatography.
4.3. Computational Details
All DFT [56] calculations were carried out using the Gaussian 16 ES64L-G16RevB.01 [57] suite of programs, and some calculated results were processed using Multiwfn [54,55]. The geometries of intermediates and transition states were optimized using the PBE0 [58,59,60] level of theory in combination with the DFT-D3 dispersion corrections with the Becke–Johnson damping scheme (D3BJ) [61,62]. The def2-SVP basis set [63,64] was used for all atoms and the solvent effects were computed with the SMD solvation model [65] (solvent = DMF). Vibrational frequency calculations were performed for all stationary points to confirm if each optimized structure was a local minimum or a transition state structure. All of the optimized transition state structures had only one imaginary (negative) frequency, and all minima (reactants, products, and intermediates) had no imaginary frequencies. The single-point energies and solvent effects were computed with the wB97XD [66]/def2-TZVPP [67,68] basis sets. The corrections of free energies [69] were applied for entropy calculations with a frequency cut-off of 100 cm^−1^ using the Shermo [70] program. The relative Gibbs free energies are given in kcal/mol, which were calculated by adding the gas-phase thermal and non-thermal corrections at 353.15 K to the single-point energies. The charge calculations were conducted at the B3LYP/6–31G + (d,p) level through becke charge [53]. The 3D images of the structures were prepared using VMD [71].
4.4. General Procedure for Control Experiments Processes
4.4.1. CuBr2 Without Other Substances Supported the Reaction
1a (1.0 mmol, 148.5 mg) and CuBr_2_ (2.0 mmol, 446.74 mg) were added to a sealed tube (40 mL). EtOH (3.0 mL) was added, and the mixture was stirred vigorously at 80 °C for 24 h. After completion, the reaction mixture was filtered. Then, the white powder and organic filtrate were obtained. XRD studies of the white powder indicated that it was CuBr (Figure S1). The organic filtrate was added into DCM (30 mL) and washed with NaCl solution (50 mL × 3). The organic mixture was dried with MgSO_4_, concentrated in vacuo, and separated with flash column chromatography (petroleum ether:ethyl acetate = 60:1) to obtain D. ^1^H NMR (500 MHz, CDCl_3_) δ 7.92 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 5.28 (q, J = 6.6 Hz, 1H), 2.42 (s, 3H), 1.89 (d, J = 6.6 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 193.0, 144.7, 131.5, 129.5, 129.1, 41.7, 21.7, 20.2.
4.4.2. The Reaction Was Supported Without CuBr
D (1.0 mmol), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to a sealed tube (40 mL). DMF (3.0 mL) was added, and the mixture was stirred vigorously at 80 °C for 24 h. After completion, the reaction mixture was added into DCM (30 mL) and washed with NaCl solution (50 mL × 3). The organic mixture was dried with MgSO_4_, concentrated in vacuo, and was separated with flash column chromatography to obtain 2a (41%).
4.4.3. CuBr Supported the Reaction
D (1.0 mmol), CuBr (1.0 mmol, 143.5 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to a sealed tube (40 mL). DMF (3.0 mL) was added, and the mixture was stirred vigorously at 80 °C for 24 h. After completion, the reaction mixture was added into DCM (30 mL) and washed with NaCl solution (50 mL × 3). The organic mixture was dried with MgSO_4_, concentrated in vacuo, and separated with flash column chromatography to obtain 2a (94%).
4.4.4. A 10% Amount of CuBr Supported the Reaction
D (1.0 mmol), CuBr (1.0 mmol, 143.5 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to a sealed tube (40 mL). DMF (3.0 mL) was added and the mixture was stirred vigorously at 80 °C for 24 h. After completion, the reaction mixture was added into DCM (30 mL) and washed with NaCl solution (50 mL × 3). The organic mixture was dried with MgSO_4_, concentrated in vacuo, and separated with flash column chromatography to obtain 2a (92%).
4.4.5. Scale-Up Experiments
1a (10.0 mmol, 1.4850 g), CuBr_2_ (20.0 mmol, 4.4670 g), TMSCF_3_ (20.0 mmol, 2.8440 g), and H_2_O (10.0 mmol, 0.1800 g) were added to a sealed tube (100 mL). DMF (30.0 mL) was added, and the mixture was stirred vigorously at 80 °C for 24 h. After completion, the reaction mixture was added into DCM (300 mL) and washed with NaCl solution (500 mL × 3). The organic mixture was dried with MgSO_4_, concentrated in vacuo, and separated with flash column chromatography to obtain 2a (63%). When the operation was changed by slowly adding CuBr, the yield of 2a increased to 92%.
4.5. General Procedure for Rectification
The α-Acyloxylation reaction of substrate 1a was carried out on a gram scale (20.2 g) under the optimized reaction conditions with TMSCF_3_ (28.4 g). After completion, the reaction mixture was added into a rectification tower, and then TMSCF_3_ was recovered (Figure S3).
4.6. Compounds Characterization
4.6.1. 1-Oxo-1-(p-tolyl)propan-2-yl Formate (2a)
According to the general procedure, 1a (1.0 mmol, 148.5 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) with a reaction time of 24 h to produce 2a (183.6 mg, 95% yield) which was isolated as a white solid powder (m.p. = 74–75 °C) after purification through silica gel chromatography (petroleum ether:ethyl acetate = 50:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.12 (s, 1H), 7.85 (d, J = 7.8 Hz, 2H), 7.29 (d, J = 7.9 Hz, 2H), 6.10 (q, J = 7.0 Hz, 1H), 2.42 (s, 3H), 1.57 (d, J = 7.0 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 195.3, 160.0, 144.7, 131.6, 129.6, 129.0, 70.6, 21.7, 17.2. IR (KBr): 2987, 2961, 2360, 2331, 1719, 1680, 1605, 1568, 1447, 1177, 1129 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_11_H_12_O_3_, 193.0865; found: 193.0868.
4.6.2. 1-(4-Ethylphenyl)-1-oxopropan-2-yl Formate (2b)
According to the general procedure, 1b (1.0 mmol, 162.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2b (165.4 mg, 80% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 90:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.12 (s, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 8.1 Hz, 2H), 6.11 (q, J = 7.0 Hz, 1H), 2.71 (q, J = 7.6 Hz, 2H), 1.57 (d, J = 7.0 Hz, 3H), 1.26 (t, J = 7.6 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 195.4, 160.1, 151.0, 131.8, 128.8, 128.4, 71.0, 29.0, 17.4, 15.1. IR (KBr): 3480, 3033, 2969, 2359, 1728, 1694, 1606, 1175, 971 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_12_H_14_O_3_, 207.1021; found: 207.1024.
4.6.3. 1-(4-Bromophenyl)-1-oxopropan-2-yl Formate (2c)
According to the general procedure, 1c (1.0 mmol, 212.0 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2c (215.1 mg, 84% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 130:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.12 (s, 1H), 7.89 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 6.04 (q, J = 7.0 Hz, 1H), 1.57 (d, J = 7.0 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 194.8, 160.0, 140.3, 132.4, 129.9, 129.2, 70.9, 17.1. IR (KBr): 3486, 3091, 2988, 2937, 1725, 1696, 1585, 1567, 1446, 1170 cm^−1^. The NMR data were in agreement with the reported results [72].
4.6.4. 1-(4-Chlorophenyl)-1-oxopropan-2-yl Formate (2d)
According to the general procedure, 1d (1.0 mmol, 168.0 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2d (175.0 mg, 83% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 160:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.12 (s, 1H), 7.89 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 6.04 (q, J = 7.0 Hz, 1H), 1.57 (d, J = 7.0 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 193.9, 159.0, 131.8, 131.2, 128.9, 128.0, 69.8, 16.1. IR (KBr): 3093, 3067, 2992, 2939, 2362, 1723, 1696, 1588, 1165, 1091 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_10_H_9_ClO_3_, 213.0318; found: 213.0318.
4.6.5. 1-(4-Fluorophenyl)-1-oxopropan-2-yl Formate (2e)
According to the general procedure, 1e (1.0 mmol,152.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2e (154.3 mg, 79% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 100:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.12 (s, 1H), 8.05–7.95 (m, 2H), 7.17 (t, J = 8.6 Hz, 2H), 6.06 (q, J = 7.0 Hz, 1H), 1.58 (d, J = 7.0 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 194.3, 166.1(d,^1^J(C,F) = 252.0 Hz), 160.0, 131.2 (d, ^3^J(C,F) = 12.6 Hz), 130.5 (d, ^4^J(C,F) = 3.0 Hz), 116.1 (d, ^2^J(C,F) = 25.2 Hz), 70.9, 17.2. IR (KBr): 3077, 2992, 2733, 2457, 1727, 1599, 1508, 1230, 1160, 971 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_10_H_9_FO_3_, 197.0614; found: 197.0618.
4.6.6. 1-(4-(Trifluoromethyl)phenyl)-1-oxopropan-2-yl Formate (2f)
According to the general procedure, 1f (1.0 mmol, 202.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2f (190.5 mg, 77% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 100:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.12 (s, 1H), 8.05 (d, J = 8.1 Hz, 2H), 7.77 (d, J = 8.1 Hz, 2H), 6.07 (q, J = 7.0 Hz, 1H), 1.59 (d, J = 7.0 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 195.2, 160.0, 137.0, 135.0 (q, ^2^J(C,F) = 25.2 Hz), 128.8, 125.9 (q, ^3^J(C,F) = 12.6 Hz), 123.4 (q, ^1^J(C,F) = 277.2 Hz), 71.1, 16.9. IR (KBr): 3503, 2994, 2943, 2360, 1730, 1619, 1580, 1412, 1325, 1169 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_11_H_9_F_3_O_3_, 247.0582; found: 247.0584.
4.6.7. 1-(4-Hydroxyphenyl)-1-oxopropan-2-yl Formate (2g)
According to the general procedure, 1g (1.0 mmol, 150.2 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2g (136.5 mg, 70% yield), which was isolated as a white solid powder (m.p. = 66–67 °C) after purification through silica gel chromatography (petroleum ether:ethyl acetate = 190:1). ^1^H NMR (400 MHz, CDCl_3_) δ 8.15 (s, 1H), 8.03 (s, 1H), 7.75 (d, J = 8.8 Hz, 2H), 6.80 (d, J = 8.8 Hz, 2H), 5.99 (q, J = 7.0 Hz, 1H), 1.48 (d, J = 7.1 Hz, 3H). ^13^C NMR (101 MHz, CDCl_3_) δ 194.6, 161.1, 159.8, 130.4, 124.8, 114.9, 70.1, 16.6. IR (KBr): 3336, 2917, 2360, 2342, 1683, 1606, 1203, 1017, 853 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_10_H_10_O_4_, 195.0657; found: 195.0655.
4.6.8. 1-Oxo-1-phenylbutan-2-yl Formate (2h)
According to the general procedure, 1h (1.0 mmol, 148.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2h (147.5 mg, 77% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 60:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.17 (s, 1H), 7.94 (d, J = 7.3 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.7 Hz, 2H), 5.98 (dd, J = 7.9, 4.2 Hz, 1H), 2.07–1.84 (m, 2H), 1.03 (t, J = 7.4 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 195.5, 160.3, 134.6, 133.7, 128.9, 128.4, 75.9, 24.8, 9.7. IR (KBr): 3064, 2975, 2939, 2359, 2342, 1730, 1697, 1597, 1580, 1449, 1223, 1174 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_11_H_12_O_3_, 193.0865; found: 193.0865.
4.6.9. 1-Oxo-1-phenylpentan-2-yl Formate (2i)
According to the general procedure, 1i (1.0 mmol, 162.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2i (156.4 mg, 76% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 65:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.16 (s, 1H), 7.95 (d, J = 7.3 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.7 Hz, 2H), 6.03 (dd, J = 7.5, 5.3 Hz, 1H), 1.91–1.84 (m, 2H), 1.57–1.43 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 195.7, 160.3, 134.5, 133.7, 128.9, 128.4, 74.7, 33.4, 18.7, 13.6. IR (KBr): 3063, 2963, 2936, 2341, 1730, 1696, 1597, 1580, 1449, 1173 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_12_H_14_O_3_, 207.1021; found: 207.1022.
4.6.10. 3-Methyl-1-oxo-1-phenylbutan-2-yl Formate (2j)
According to the general procedure, 1j (1.0 mmol, 162.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2j (136.0 mg, 66% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 90:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.20 (s, 1H), 7.95 (d, J = 7.4 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.7 Hz, 2H), 5.90 (d, J = 4.3 Hz, 1H), 2.39–2.29 (m, 1H), 1.07 (d, J = 6.8 Hz, 3H), 0.94 (d, J = 6.8 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 195.7, 160.5, 135.3, 133.6, 128.9, 128.4, 78.9, 30.2, 19.5, 16.7. IR (KBr): 2963, 2360, 2341, 1730, 1696, 1260, 1094, 1019, 799 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_12_H_14_O_3_, 207.1021; found: 207.1023.
4.6.11. 1-Oxo-1-phenylhexan-2-yl Formate (2k)
According to the general procedure, 1k (1.0 mmol, 176.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2k (165.8 mg, 75% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 85:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.16 (s, 1H), 7.95 (d, J = 7.8 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.6 Hz, 2H), 6.01 (dd, J = 8.6, 4.1 Hz, 1H), 1.97–1.79 (m, 2H), 1.47–1.42 (m, 2H), 1.40–1.33 (m, 2H), 0.89 (t, J = 7.2 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 195.6, 160.3, 134.5, 133.7, 128.8, 128.4, 74.8, 31.1, 27.4, 22.2, 13.8. IR (KBr): 3064, 2959, 2932, 2872, 1728, 1697, 1598, 1581, 1449, 1171 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_13_H_16_O_3_, 221.1178; found: 221.1176.
4.6.12. 2-Methyl-1-oxo-1-phenylpropan-2-yl Formate (2l)
According to the general procedure, 1l (1.0 mmol, 148.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2l (134.6 mg, 70% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 110:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.01 (d, J = 7.1 Hz, 2H), 7.86 (s, 1H), 7.51 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.8 Hz, 2H), 1.76 (s, 6H). ^13^C NMR (126 MHz, CDCl_3_) δ 198.5, 160.0, 134.1, 132.7, 128.8, 128.4, 84.9, 25.4. IR (KBr): 3063, 2995, 2943, 2353, 1970, 1722, 1685, 1598, 1467, 1385, 1280, 1137, 713 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_11_H_12_O_3_, 193.0865; found: 193.0864.
4.6.13. 2-Oxo-1,2-diphenylethyl Formate (2m)
According to the general procedure, 1m (1.0 mmol, 196.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2m (170.9 mg, 71% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 95:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.21 (s, 1H), 7.93 (d, J = 7.3 Hz, 2H), 7.52–7.45 (m, 3H), 7.42–7.33 (m, 5H), 7.00 (s, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 192.7, 160.0, 134.3, 133.7, 133.1, 129. 6, 129.2, 128.8, 128.7, 77.1. IR (KBr): 2917, 2849, 2360, 1733, 1697, 1472, 1261, 1151, 803 cm^−1^. The NMR data were in agreement with the reported results [73].
4.6.14. 2-Oxo-2-(o-tolyl)ethyl Formate (2n)
According to the general procedure, 1n (1.0 mmol, 134.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2n (89.7 mg, 50% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 100:1). ^1^H NMR (400 MHz, CDCl_3_) δ 8.16 (s, 1H), 7.57–7.50 (m, 1H), 7.41–7.32 (m, 1H), 7.23 (d, J = 7.5 Hz, 2H), 5.21 (s, 2H), 2.45 (s, 3H). ^13^C NMR (101 MHz, CDCl_3_) δ 193.4, 159.0, 138.2, 133.1, 131.3, 127.0, 124.7, 65.5, 28.7, 20.1. IR (KBr): 2934, 2361, 2341, 1733, 1699, 1457, 1161, 934, 823 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_10_H_10_O_3_, 179.0708; found: 179.0708.
4.6.15. 2-Oxo-2-(m-tolyl)ethyl Formate (2o)
According to the general procedure, 1o (1.0 mmol, 134.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2o (93.1 mg, 52% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 90:1). ^1^H NMR (400 MHz, CDCl_3_) δ 8.15 (s, 1H), 7.63 (s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 5.33 (s, 2H), 2.31 (s, 3H). ^13^C NMR (101 MHz, CDCl_3_) δ 190.3, 159.0, 137.8, 133.8, 132.9, 127.7, 127.2, 123.9, 64.4, 20.3. IR (KBr): 3439, 3005, 2363, 1726, 1694, 1607, 1427, 1190, 795 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_10_H_10_O_3_, 179.0708; found: 179.0708.
4.6.16. 2-Oxo-2-(p-tolyl)ethyl Formate (2p)
According to the general procedure, 1p (1.0 mmol, 134.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2p (98.1 mg, 55% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 110:1). ^1^H NMR (400 MHz, CDCl_3_) δ 8.18 (s, 1H), 7.75 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 5.35 (s, 2H), 2.36 (s, 3H). ^13^C NMR (101 MHz, CDCl_3_) δ 189.6, 159.1, 144.1, 130.4, 128.6, 126.9, 64.2, 20.8. IR (KBr): 3440, 2918, 2360, 1727, 1607, 1422, 1282, 1158, 815 cm^−1^. The NMR data were in agreement with the reported results [74].
4.6.17. 1-(5-Methylfuran-2-yl)-1-oxopropan-2-yl Formate (2q)
According to the general procedure, 1q (1.0 mmol, 138.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2q (119.4 mg, 66% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 100:1).^1^H NMR (500 MHz, CDCl_3_) δ 8.12 (s, 1H), 7.25 (d, J = 3.6 Hz, 1H), 6.22 (d, J = 3.5 Hz, 1H), 5.86 (q, J = 6.9 Hz, 1H), 2.42 (s, 3H), 1.58 (d, J = 7.0 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 183.8, 160.0, 158.8, 148.9, 120.9, 109.4, 71.0, 17.4, 14.1. IR (KBr): 3734, 2939, 2360, 2341, 1716, 1669, 1514, 1209, 1168, 1129 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_9_H_10_O_4_, 183.0657; found: 183.0655.
4.6.18. 1-Oxo-1-(thiophen-2-yl)propan-2-yl Formate (2r)
According to the general procedure, 1r (1.0 mmol, 140.3 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2r (121.1 mg, 66% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 110:1). ^1^H NMR (500 MHz, CDCl_3_) δ 8.13 (s, 1H), 7.82 (d, J = 3.0 Hz, 1H), 7.72 (d, J = 4.9 Hz, 1H), 7.19–7.16 (m, 1H), 5.90 (q, J = 6.9 Hz, 1H), 1.63 (d, J = 7.1 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 188.7, 159.9, 140.3, 134.8, 132.8, 128.3, 71.8, 17.8. IR (KBr): 3101, 2992, 2359, 1726, 1675, 1517, 1414, 1242, 1175 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_8_H_8_O_3_S, 185.0272; found: 185.0273.
4.6.19. 1-Oxo-2,3-dihydro-1H-inden-2-yl Formate (2s)
According to the general procedure, 1s (1.0 mmol, 132.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2s (97.2 mg, 55% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 150:1). ^1^H NMR (400 MHz, CDCl_3_) δ 8.15 (s, 1H), 7.72 (d, J = 7.7 Hz, 1H), 7.58 (t, J = 7.3 Hz, 1H), 7.42–7.32 (m, 2H), 5.51–5.43 (m, 1H), 3.62 (dd, J = 17.0, 8.1 Hz, 1H), 3.01 (dd, J = 17.0, 4.9 Hz, 1H). ^13^C NMR (101 MHz, CDCl_3_) δ 198.7, 159.1, 149.2, 135.1, 133.2, 127.3, 125.6, 123.5, 72.4, 32.2. IR (KBr): 3431, 2931, 2360, 1715, 1608, 1466, 1155, 997, 885 cm^−1^. The NMR data were in agreement with the reported results [75].
4.6.20. 1-Oxo-1,2,3,4-tetrahydronaphthalen-2-yl Formate (2t)
According to the general procedure, 1t (1.0 mmol, 146.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMF (3.0 mL) to produce 2t (95.2 mg, 50% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 110:1). ^1^H NMR (400 MHz, CDCl_3_) δ 8.18 (s, 1H), 7.94 (d, J = 7.8 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.28–7.17 (m, 2H), 5.56 (dd, J = 13.4, 5.2 Hz, 1H), 3.20–2.97 (m, 2H), 2.40–2.16 (m, 2H). ^13^C NMR (101 MHz, CDCl_3_) δ 191.1, 158.9, 142.0, 133.1, 130.3, 127.7, 126.8, 126.0, 73.1, 28.0, 26.8. The NMR data were in agreement with the reported results [75].
4.6.21. 1-Oxo-1-(p-tolyl)propan-2-yl Acetate (3a)
According to the general procedure, 1a (1.0 mmol, 148.5 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMA (3.0 mL) to produce 3a (182.0 mg, 88% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 65:1). ^1^H NMR (500 MHz, CDCl_3_) δ 7.84 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 5.95 (q, J = 7.0 Hz, 1H), 2.41 (s, 3H), 2.14 (s, 3H), 1.52 (d, J = 7.1 Hz, 3H).^13^C NMR (126 MHz, CDCl_3_) δ 196.4, 170.4, 144.5, 131.8, 129.5, 128.6, 71.4, 21.7, 20.7, 17.3. The NMR data were in agreement with the reported results [76].
4.6.22. 1-(4-Bromophenyl)-1-oxopropan-2-yl Benzoate (3c)
According to the general procedure, 1c (1.0 mmol, 212.0 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMA (3.0 mL) to produce 3c (215.2 mg, 80% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 160:1). ^1^H NMR (500 MHz, CDCl_3_) δ 7.80 (d, J = 8.6 Hz, 2H), 7.62 (d, J = 8.6 Hz, 2H), 5.88 (q, J = 7.0 Hz, 1H), 2.14 (s, 3H), 1.51 (d, J = 7.0 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 196.0, 170.4, 133.2, 132.1, 129.9, 128.8, 71.3, 20.7, 17.0. The NMR data were in agreement with the reported results [76].
4.6.23. 1-Oxo-1-phenylpentan-2-yl Acetate (3i)
According to the general procedure, 1i (1.0 mmol, 162.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMA (3.0 mL) to produce 3i (164.5 mg, 75% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 35:1). ^1^H NMR (500 MHz, CDCl_3_) δ 7.94 (d, J = 7.2 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.8 Hz, 2H), 5.88 (dd, J = 7.7, 5.1 Hz, 1H), 2.16 (s, 3H), 1.89–1.79 (m, 2H), 1.55–1.39 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 196.7, 170.7, 134.9, 133.5, 128.8, 128.4, 75.2, 33.4, 20.7, 18.9, 13.7. The NMR data were in agreement with the reported results [76].
4.6.24. 1-Oxo-1-phenylhexan-2-yl Acetate (3k)
According to the general procedure, 1k (1.0 mmol, 176.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMA (3.0 mL) to produce 3k (174.1 mg, 74% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 85:1). ^1^H NMR (500 MHz, CDCl_3_) δ 7.94 (d, J = 7.0 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.8 Hz, 2H), 5.86 (dd, J = 8.6, 4.2 Hz, 1H), 2.16 (s, 3H), 2.03–1.62 (m, 2H), 1.50–1.20 (m, 4H), 0.89 (t, J = 7.2 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 196.7, 170.7, 134.9, 133.5, 128.8, 128.4, 75.4, 31.1, 27.7, 22.3, 20.7, 13.8. IR (KBr): 2962, 2933, 2874, 2364, 1740, 1696, 1597, 1448, 1375, 1230, 697 cm^−1^. The NMR data were in agreement with the reported results [77].
4.6.25. 2-Oxo-1,2-diphenylethyl Acetate (3m)
According to the general procedure, 1m (1.0 mmol, 196.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMA (3.0 mL) to produce 3m (178.2 mg, 70% yield), which was isolated as a white solid powder (m.p. = 72–73 °C) after purification through silica gel chromatography (petroleum ether:ethyl acetate = 110:1). ^1^H NMR (500 MHz, CDCl_3_) δ 7.95–7.91 (m, 2H), 7.52–7.46 (m, 3H), 7.41–7.33 (m, 5H), 6.86 (s, 1H), 2.20 (s, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 193.8, 170.5, 134.7, 133.7, 133.5, 129.3, 129.2, 128.8, 128.7, 128.7, 77.7, 20.8. The NMR data were in agreement with the reported results [77].
4.6.26. 1-(5-Methylfuran-2-yl)-1-oxopropan-2-yl Acetate (3q)
According to the general procedure, 1q (1.0 mmol, 138.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMA (3.0 mL) to produce 3q (116.9 mg, 60% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 150:1). ^1^H NMR (500 MHz, CDCl_3_) δ 7.22 (d, J = 3.5 Hz, 1H), 6.19 (d, J = 3.4 Hz, 1H), 5.70 (q, J = 7.0 Hz, 1H), 2.41 (s, 3H), 2.14 (s, 3H), 1.53 (d, J = 7.0 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 184.8, 170.3, 158.5, 149.1, 120.6, 109.3, 71.4, 20.7, 17.3, 14.1. IR (KBr): 3122, 2992, 2360, 2341, 1742, 1682, 1515, 1372, 1238, 1040 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_10_H_12_O_4_, 197.0814; found: 197.0812.
4.6.27. 1-Oxo-1-(thiophen-2-yl)propan-2-yl Acetate (3r)
According to the general procedure, 1r (1.0 mmol, 140.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMA (3.0 mL) to produce 3r (119.6 mg, 60% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 100:1). ^1^H NMR (500 MHz, CDCl_3_) δ 7.81 (d, J = 4.6 Hz, 1H), 7.70 (d, J = 5.7 Hz, 1H), 7.19–7.13 (m, 1H), 5.74 (q, J = 7.0 Hz, 1H), 2.16 (s, 3H), 1.58 (d, J = 7.1 Hz, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 189.7, 170.3, 140.5, 134.4, 132.6, 128.2, 72.4, 20.8, 17.7. IR (KBr): 3091, 2964, 2928, 2328, 1741, 1669, 1517, 1448, 1413, 1235, 1080, 798 cm^−1^. The NMR data were in agreement with the reported results [76].
4.6.28. 2-Oxo-2-phenylethyl Acetate (3u)
According to the general procedure, 1u (1.0 mmol, 120.2 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) were added to DMA (3.0 mL) to produce 3u (137.6 mg, 77% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 90:1). ^1^H NMR (500 MHz, CDCl_3_) δ 7.91 (d, J = 7.3 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.7 Hz, 2H), 5.34 (s, 2H), 2.22 (s, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 192.2, 170.4, 134.2, 133.9, 128.9, 127.8, 66.0, 20.5. The NMR data were in agreement with the reported results [78].
4.6.29. 1-Oxo-1-(p-tolyl)propan-2-yl Benzoate (4a)
According to the general procedure, the combination of 1a (1.0 mmol, 148.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), H_2_O (1.0 mmol, 18.0 mg), and N,N-dimethylbenzamide (3 g) produced 4a (185.5 mg, 70% yield), which was isolated as a white solid powder (m.p. = 67–68 °C) after purification through silica gel chromatography (petroleum ether:ethyl acetate = 190:1). ^1^H NMR (400 MHz, CDCl_3_) δ 7.98 (d, J = 7.0 Hz, 2H), 7.79 (d, J = 8.3 Hz, 2H), 7.43 (t, J = 7.5 Hz, 1H), 7.30 (t, J = 7.8 Hz, 2H), 7.14 (d, J = 8.0 Hz, 2H), 6.07 (q, J = 7.0 Hz, 1H), 2.26 (s, 3H), 1.54 (d, J = 7.0 Hz, 3H). ^13^C NMR (101 MHz, CDCl_3_) δ 195.1, 164.9, 143.4, 132.2, 130.8, 128.8, 128.5, 128.4, 127.6, 127.3, 70.8, 20.6, 16.2. The NMR data were in agreement with the reported results [79].
Crystal Data for C_17_H_16_O_3_ (M =268.30 g/mol): monoclinic, space group P2_1_/c (no. 14), a = 20.4570(9) Å, b = 9.1734(4) Å, c = 16.0710(7) Å, β = 109.656(5)°, V = 2840.1(2) Å^3^, Z = 8, T = 123.15 K, μ (MoKα) = 0.085 mm^−1^, Dcalc = 1.255 g/cm^3^, 29584 reflections measured (4.228° ≤ 2Θ ≤ 52.744°), with 5809 unique (Rint = 0.0506, R_sigma_ = 0.0337) which were used in all of the calculations. The final R1 was 0.0443 (I > 2σ(I)) and the wR2 was 0.1143 (all data).
4.6.30. 2-Oxopentan-3-yl Benzoate (4v)
According to the general procedure, the combination of 1v (1.0 mmol, 86.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), H_2_O (1.0 mmol, 18.0 mg), and N,N-dimethylbenzamide (3 g) produced 4v (113.0 mg, 55% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 200:1). ^1^H NMR (400 MHz, CDCl_3_) δ 8.02 (d, J = 7.0 Hz, 2H), 7.52 (t, J = 7.5 Hz, 1H), 7.39 (t, J = 7.7 Hz, 2H), 5.11 (dd, J = 7.7, 4.8 Hz, 1H), 2.14 (s, 3H), 1.99–1.78 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). ^13^C NMR (101 MHz, CDCl_3_) δ 204.5, 165.1, 132.4, 128.7, 128.4, 127.5, 79.1, 25.3, 22.9, 8.6. IR (KBr): 3065, 2973, 2361, 1717, 1602, 1452, 1271, 1177, 1110, 711 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_12_H_14_O_3_, 207.1021; found: 207.1022.
4.6.31. 2-Oxohexan-3-yl Benzoate (4w)
According to the general procedure, the combination of 1w (1.0 mmol, 100.1 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), H_2_O (1.0 mmol, 18.0 mg), and N,N-dimethylbenzamide (3 g) produced 4w (102.8 mg, 50% yield) which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 200:1).^1^H NMR (400 MHz, CDCl_3_) δ 7.99 (d, J = 7.1 Hz, 2H), 7.48 (t, J = 7.4 Hz, 1H), 7.36 (t, J = 7.7 Hz, 2H), 5.14 (t, J = 6.4 Hz, 1H), 2.11 (s, 3H), 1.82–1.73 (m, 2H), 1.48–1.35 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H). ^13^C NMR (101 MHz, CDCl_3_) δ 204.5, 165.0, 132.4, 128.7, 128.4, 127.5, 78.0, 31.5, 25.1, 17.6, 12.7. IR (KBr): 2962, 2875, 2361, 1719, 1602, 1452, 1277, 1113, 1071, 1027, 713 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_13_H_16_O_3_, 221.1178; found: 221.1176.
4.6.32. 1-Oxo-1-(p-tolyl)propan-2-yl Propionate (5a)
According to the general procedure, the combination of 1a (1.0 mmol, 148.5 mg), CuBr_2_ (2.0 mmol, 446.7 mg), TMSCF_3_ (2.0 mmol, 284.4 mg), and H_2_O (1.0 mmol, 18.0 mg) N,N-dimethylpropionamide (3.0 mL) produced 5a (175.7 mg, 80% yield), which was isolated as a colorless oil after purification through silica gel chromatography (petroleum ether:ethyl acetate = 50:1). ^1^H NMR (400 MHz, CDCl_3_) δ 7.75 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 5.87 (q, J = 7.0 Hz, 1H), 2.38–2.33 (m, 2H), 2.31 (s, 3H), 1.42 (d, J = 7.1 Hz, 3H), 1.06 (t, J = 7.6 Hz, 3H). ^13^C NMR (101 MHz, CDCl_3_) δ 195.5, 172.8, 143.4, 130.8, 128.4, 127.5, 70.1, 26.3, 20.6, 16.2, 7.9. IR (KBr): 3034, 2985, 2940, 2359, 2330, 1738, 1694, 1606, 1572, 1183 cm^−1^. HRMS-ESI (m/z): [M + H]^+^ calcd for C_13_H_16_O_3_, 221.1178; found: 221.1176.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tietze L.F. Domino reactions in organic synthesis Chem. Rev.19969611513610.1021/cr 950027 e 11848746 · doi ↗ · pubmed ↗
- 2Nicolaou K.C. Edmonds D.J. Bulger P.G. Cascade reactions in total synthesis Angew. Chem. Int. Ed.2006457134718610.1002/anie.20060187217075967 · doi ↗ · pubmed ↗
- 3Grondal C. Jeanty M. Enders D. Organocatalytic cascade reactions as a new tool in total synthesis Nat. Chem.2010216717810.1038/nchem.53921124474 · doi ↗ · pubmed ↗
- 4Volla C.M.R. Atodiresei I. Rueping M. Catalytic C–C bond-forming multi-component cascade or domino reactions: Pushing the boundaries of complexity in asymmetric organocatalysis Chem. Rev.20141142390243110.1021/cr 400215 u 24304297 · doi ↗ · pubmed ↗
- 5Ardkhean R. Caputo D.F.J. Morrow S.M. Shi H. Xiong Y. Anderson E.A. Cascade polycyclizations in natural product synthesis Chem. Soc. Rev.2016451557156910.1039/C 5CS 00105 F 26791791 · doi ↗ · pubmed ↗
- 6Chauhan P. Mahajan S. Enders D. Achieving Molecular Complexity via Stereoselective Multiple Domino Reactions Promoted by a Secondary Amine Organocatalyst Acc. Chem. Res.2017502809282110.1021/acs.accounts.7b 0040629125741 · doi ↗ · pubmed ↗
- 7Zhang M. Gong Y. Zhou W. Zhou Y. Liu X.-L. Recent advances of chromone-based reactants in the catalytic asymmetric domino annulation reaction Org. Chem. Front.202183968398910.1039/D 1QO 00269 D · doi ↗
- 8Okiko Miyata T.M. Masafumi Ueda, Umpolung reactions at the α-carbon position of carbonyl compounds ARKIVOC 20132013608110.3998/ark.5550190.0014.20721246693 · doi ↗ · pubmed ↗