A Novel Pathogenic Variant of DICER1 Gene in a Young Greek Patient with 2 Different Sex-Cord Ovarian Tumors and Multinodular Goiter
Afroditi Roumpou, Argyro-Ioanna Ieronimaki, Aspasia Manta, Ioannis G. Panayiotides, Constantine A. Stratakis, Sophia Kalantaridou, Melpomeni Peppa

TL;DR
A young Greek patient with two ovarian tumors and a goiter was found to have a new mutation in the DICER1 gene, which is linked to increased cancer risk.
Contribution
A novel pathogenic variant in the DICER1 gene is reported in association with multiple tumors and multinodular goiter in a young patient.
Findings
A new DICER1 gene variant was identified in a young patient with multinodular goiter and ovarian tumors.
The case highlights the importance of considering DICER1 mutations in young patients with MNG and other neoplasms.
SLCT and JGCT are rare tumor types associated with DICER1 syndrome, and this case adds to the clinical spectrum.
Abstract
DICER1 syndrome (DICERs) represents a tumor predisposition genetic syndrome, inherited in an autosomal dominant manner. Germline loss-of-function variants of the DICER1 gene lead to impaired processing of microRNA, gene expression, and increased risk of tumorigenesis. Although pleuropulmonary blastoma (PPB) is the hallmark of the syndrome, multiple extrapulmonary malignant and non-malignant conditions have also been described, including multinodular goiter (MNG) and sex-cord stromal tumors. MNG is one of the most common components and is associated with an increased risk of thyroid carcinoma. Sertoli–Leydig cell tumor (SLCT) represents the most prevalent type of sex-cord stromal tumor associated with the syndrome, whereas juvenile granulosa cell tumor (JGCT) is considered to be a very rare phenotype. They both may present with abdominal pain due to mass effect and menstrual…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
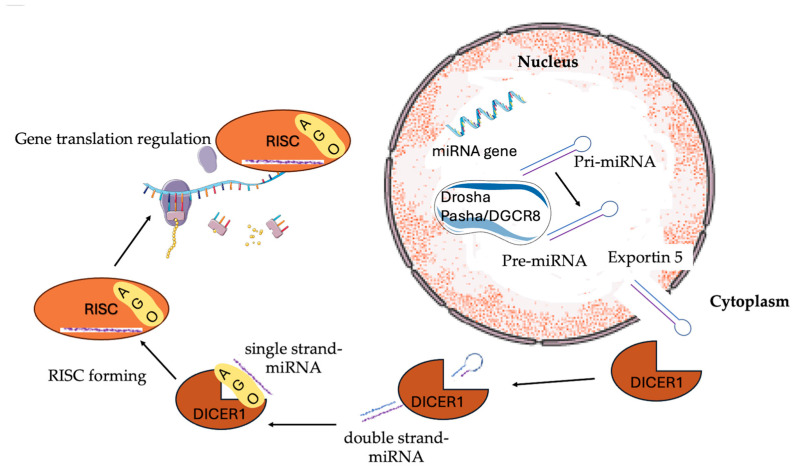 Figure 1
Figure 1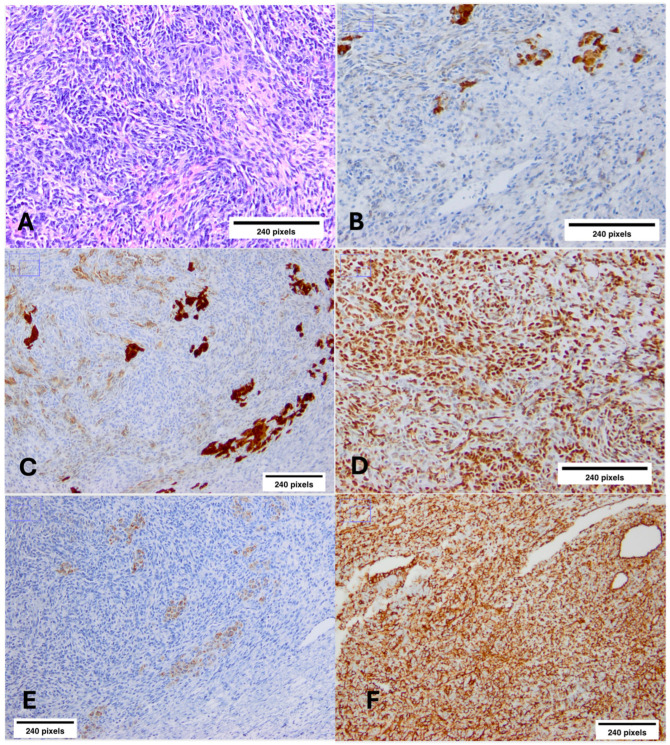 Figure 2
Figure 2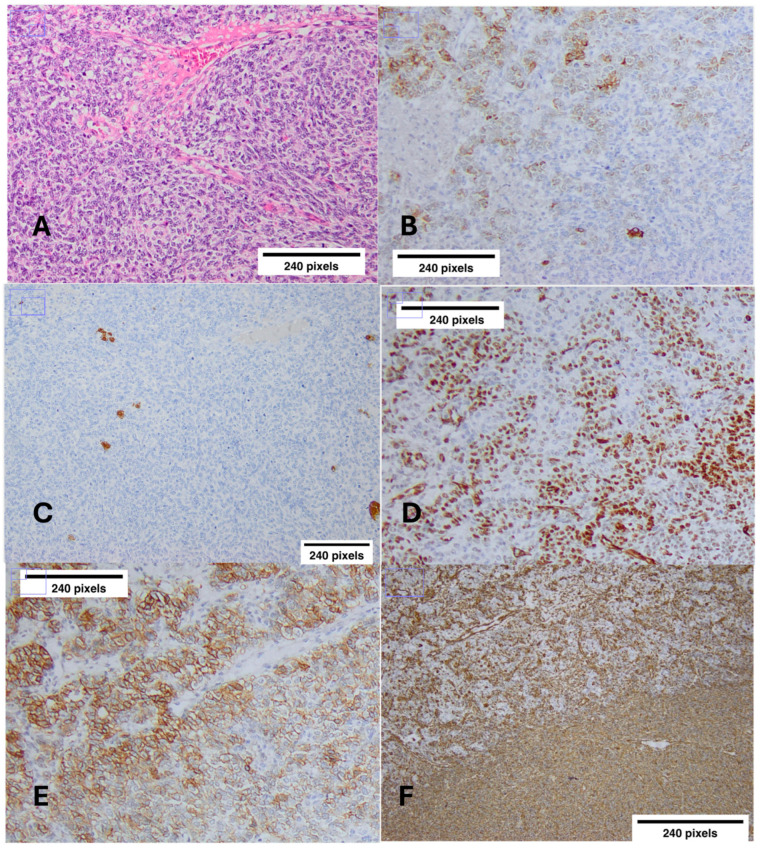 Figure 3
Figure 3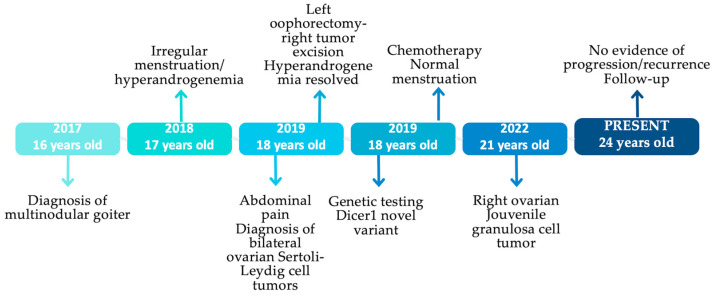 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital Diaphragmatic Hernia Studies · Neuroendocrine Tumor Research Advances · Particle accelerators and beam dynamics
1. Introduction
DICER1 syndrome (DICER1s) is a genetic syndrome characterized by the development of multiple benign and malignant neoplasms, as well as non-neoplastic conditions. A plethora of germline loss-of-function alterations in the homonymous gene results in impaired microRNA (miRNA) processing, altered gene expression, and tumorigenesis [1]. The DICER1 gene is located at 14q32.13, and it encodes a multidomain endoribonuclease of 1922 amino acids that plays a pivotal role in the synthesis pathway of miRNA [2]. Specifically, RNA polymerase II transcribes microRNAs from pri-miRNAs, which are long RNA precursors [3]. Pri-miRNAs are processed in the nucleus by the RNA-binding protein Pasha/DGCR85 (DiGeorge critical region 8) and the RNase III enzyme Drosha to form pre-miRNAs that are folded into stem-loop structures [4,5]. After exporting the nucleus by exportin 5 [6], 3p and 5p miRNAs are cleaved from the miRNA precursor by the domains IIIa and IIIb of the ribonuclease DICER1 and, subsequently, produce the miRNA, a double-stranded RNA [7,8]. Additionally, DICER1 promotes the formation of the RNA-induced silencing complex (RISC), which includes other proteins, such as members of the Argonaute (AGO) protein family. One of the two complementary short RNAs gets incorporated into the RISC complex, which targets and regulates messenger RNA, by either promoting its destruction or disrupting its translation (Figure 1) [9,10,11,12].
DICER1s is caused by germline inactivating alterations in the DICER1 gene, inherited with an autosomal dominant pattern, while a noteworthy percentage of all cases (10–20%) seem to arise de novo [13]. Deletions, duplications, insertions, transitions, and transversions are among the mutations found in DICER1 [14]. The majority of patients developing DICER1-associated tumors, except for one hereditary germline DICER1 variant, also have another acquired somatic missense DICER1 alteration in one of the “hot-spot” codons included in the RNAse IIIb domain (E1813, D1705, D1709, D1713, and G1809) [2]. Moreover, approximately 10% of people with a predisposing DICER1 variant are mosaic, meaning the somatic mutations are acquired during postzygotic development. Mosaic individuals are rather uncommon, but their detection poses a special challenge for germline testing, as only a portion of cells harbor the mutation [15,16]. It is considered that approximately 1:2500–1:10,600 individuals in the general population are heterozygotes for a pathogenic or likely pathogenic DICER1 variant [17].
Since the first description of pleuropulmonary blastoma (PPB) in childhood by Manivel et al. [18], the term “pleuropulmonary blastoma familial tumor susceptibility syndrome” has been introduced until the identification of DICER1 gene pathogenic variants as the underlying cause of the disorder [19]. DICERs may present with a vast variety of clinical phenotypes apart from PPB, including malignant tumors, such as blastomas (pituitary, pineal), ovarian Sertoli–Leydig cell tumor (SLCT), cervical embryonal rhabdomyosarcoma, Wilms tumor, renal sarcoma, thyroid carcinoma, mesenchymal hamartoma of the liver, and neuroblastoma [13,20,21,22]. The syndrome also involves some benign conditions, such as multinodular goiter (MNG), cystic nephroma, hamartomatous intestinal polyps, and nasal chondromesenchymal hamartoma [23,24]. Non-neoplastic disorders such as macrocephaly, kidney structural abnormalities, retinal abnormalities, dental perturbations, and the GLOW syndrome (global developmental delay, lung cysts, overgrowth, and Wilms tumor) have also been observed in people with germline DICER1 variants [19]. Although they are at a higher risk of developing cancer, the majority of carriers with a germline DICER1 variant may have healthy lives [10]. The conditions associated with DICERs are categorized regarding frequency and malignant potential in Table 1 and Table 2, respectively [25].
PPB is the most frequent manifestation and the main cause of mortality, while ovarian SLCT, MNG, and cystic nephroma are some of the most prevalent components [14,24,26]. Early-onset MNG has been strongly related to the syndrome. In fact, it has been reported that the incidence of MNG or thyroidectomy among carriers of DICER1 germline pathogenic variants is 75% and 17% in women and men, respectively, before the age of 40 years [27]. In DICER1-related MNG, molecular analysis of the nodules has indicated that they are clonal, since they harbor a second somatic mutation, different for each nodule, in addition to the responsible germline alteration [2,28]. The risk for thyroid carcinoma is increased by over 16 times compared to healthy controls, with minimally invasive follicular thyroid carcinoma and the follicular variant of papillary thyroid carcinoma being the most frequently reported [27]. Generally, thyroid cancer is associated with a favorable prognosis, similar to that of sporadic differentiated thyroid carcinoma [13].
Neoplasms of the gynecologic tract, especially ovarian SLCTs, could be the first clinical manifestation of DICER1s running in a family and usually develop from childhood until adulthood [16]. SLCTs are an extremely rare type of ovarian sex-cord stromal tumors (about 1%) [29], accounting for less than 0.5% of all primary ovarian neoplasms [30,31]. However, SLCTs constitute the second most common tumors associated with DICER1s, after PPBs [32,33,34,35,36,37], with more than 50% of SLCT patients carrying a pathogenic variant [38]. They may be well, moderately, or poorly differentiated according to the World Health Organization (WHO) criteria [39]. Most SLCTs are well differentiated, have a favorable prognosis, and have rare recurrence, whereas less differentiated tumors may have a more aggressive disease course [29,40]. Although even advanced-stage SLCTs can have a positive prognosis due to their susceptibility to chemotherapy, 20% recur or develop potentially lethal metastases [41]. It has been supported that 97–100% of patients with intermediate or poorly differentiated SLCTs have a DICER1 pathogenic variant, while this is noted in only 12% of those with well differentiated tumors [1,33,42]. Compared to sporadic SLCTs, patients with DICER1-associated SLCTs tend to have features associated with hyperandrogenemia due to hormone production, early presentation, and increased risk of recurrence [35,37]. The mortality rate with SLCTs in DICER1s is minimal, and fewer than 5% of recorded deaths are linked to SLCTs [10].
Granulosa cell tumors account for 1–5% of all ovarian tumors, arise from the granulosa cells of the ovarian follicle, and are divided into adult and juvenile types, with the latter being the minority of the cases, presenting in younger patients, usually <30 years old [43,44,45]. Clinical presentation usually includes abdominal pain and increasing abdominal girth, and in cases of hormonally active tumors (estradiol production), menstrual irregularities, or precocious puberty. Patients with juvenile granulosa cell tumors (JGCT) have a very stage-dependent prognosis, with a 97% survival rate in patients with stage 1 tumors that are limited to the ovary [16]. Herein, we present for the first time a case of a young Greek female patient with a history of non-toxic MNG and both a bilateral SLCT diagnosed at the age of 18 years and JGCT at the age of 21 years, due to a novel pathogenic DICER1 gene variant.
2. Case Presentation
2.1. Multinodular Goiter (MNG)
In 2017, a 16-year-old Greek female patient was referred to our Endocrinology Unit for further investigation of an MNG found incidentally on thyroid ultrasonography. In particular, the imaging identified 7 nodules ranging from 5.5 to 25 mm in size. Fine needle aspiration (FNA) of the two larger nodules was reported as TBSII (The Bethesda system), according to the Bethesda classification system, being consistent with nodular thyroid hyperplasia. Thyroid function tests and calcitonin levels (3.3 ng/L, Ref: 1–10 ng/L) were unremarkable, and she was advised to regular follow-up.
2.2. Irregular Menstruation-Hyperandrogenemia
One year later (2018), at the age of 17 years, she was referred to a gynecologist due to irregular menstruation and acne. Menarche occurred at the age of 12 years, and until then, she did not mention any menstrual irregularities. Physical examination revealed normal breast and pubic hair development (Tanner stage V). The hormone profile revealed raised testosterone levels (3.1 ng/mL, Ref: 0.1–0.5 ng/mL), low follicle-stimulating hormone (FSH; 1.3 mIU/mL, Ref: 3–8.1 mIU/mL), and low normal luteinizing hormone (LH; 2.3 mIU/mL, Ref: 1.8–11.8 mIU/mL) levels. SHBG (sex hormone-binding globulin), DHEA-S (dehydroepiandrosterone sulfate), androstenedione, 17-hydroxyprogesterone, prolactin, and estradiol levels were all within the normal limits. We have no data regarding pelvic ultrasound findings at that time. Hyperandrogenemia and irregular menstruation were attributed to polycystic ovary syndrome by her doctor, and she was advised to take oral contraceptive pills, which she stopped after a month due to pill dysphagia.
2.3. Bilateral Ovarian Sertoli–Leydig Cell Tumors (SLCTs)
In 2019, at the age of 18, she presented to the emergency department with bloating and left-sided abdominal pain. Computed tomography (CT) imaging of the abdomen and pelvis showed a massive mass (20.6 cm in its largest dimension), occupying most of the left side of the abdomen, possibly arising from the left ovary, causing severe obstructive phenomena of the iliac vessels and both the ureters, and resulting in significant ascites. Another smaller mass (5.6 cm in its largest dimension) arising from the right ovary was also depicted. Complementary imaging with abdominal magnetic resonance imaging (MRI) and CT of the thorax revealed no evidence of metastatic disease to the abdomen, pelvis, or chest, but showed small bilateral pleural effusions, suggesting Meigs syndrome (pleural effusion, ascites, and benign ovarian fibroma) [46].
The patient was referred to the gynecology department of our hospital, and the findings were further confirmed by transvaginal and pelvic ultrasonography. She immediately underwent unilateral left oophorectomy and tumor excision of the right ovary. Histology showed both lesions to be moderately differentiated SLCTs. Tumor cells were immunostained for CKAE1/AE3, inhibin, calretinin, WT-1, and focally for melan-A. Any immunohistochemistry (IHC) was indicative of the presence or absence of these proteins, as a quantification method [e.g., immunoblots (Western) for protein quantification] was not performed (Figure 2). No capsular tearing was documented; 15 left parametrial lymph nodes were tumor-free. Since cytologic examination of ascitic fluid was negative for malignant cells, tumor stage was IB according to The International Federation of Gynecology and Obstetrics (FIGO) staging system.
Laboratory investigation of tumor markers showed that cancer antigen 125 (Ca 125) and inhibin were markedly elevated (359.7, Ref: <35 U/mL and 443, Ref: 2–80 pg/mL, respectively), whereas alpha-fetoprotein (AFP; 13.7, Ref: <40 ng/mL), carcinoembryonic antigen (CEA; 0.3, Ref: <2.5 ng/mL), and cancer antigen 15–3 (Ca 15–3; 15.4, Ref: ≤30 U/mL) were normal. Notably, Ca 125 and inhibin decreased to normal approximately 20 days after surgery, as did the hormone profile, with testosterone levels returning to normal (0.4 ng/mL, Ref: 0.05–0.52 ng/mL) (Table 3). Following surgery, the patient was referred to the medical oncology department, where she was advised to receive adjuvant chemotherapy after completing fertility preservation therapy. The latter failed due to unsuccessful ovarian stimulation. FIGO stage, intraperitoneal tumor rupture, and possibly tumor size (>5 cm) are significant prognostic factors [47,48]. Due to the FIGO stage (>IA) and the tumor size (20.6 cm), and according to European Society for Medical Oncology clinical practice guidelines, our patient received three cycles of chemotherapy in total (BEP regimen; bleomycin–etoposide–cisplatin) [41]. Her menstruation cycle returned to normal after the completion of chemotherapy sessions.
2.4. Jouvenile Granulosa Cell Tumor (JGCT)
Three years postoperatively (2022, 21 years old), during follow-up imaging with abdominal MRI, a large multilocular cystic lesion 13 × 9 × 7.5 cm at the right parametrial area was depicted. Histology showed the excised lesion to be a JGCT; tumor cells were immunostained for CKAE1/AE3 (focally in a “dot-like” juxtanuclear pattern), vimentin, inhibin, CD56, CD99, WT-1, PR, and focally for calretinin (again, any IHC was indicative of the presence or absence of these proteins) (Figure 3). No capsular tearing was documented; seven right pelvic lymph nodes were tumor free. Tumor stage was IA according to the FIGO staging system. Stage IA granulosa cell tumors have an excellent prognosis after surgery alone and do not require adjuvant therapy according to European Society for Medical Oncology clinical practice guidelines [41]. Therefore, the patient was referred to the medical oncology department for follow-up.
2.5. Genetic Testing
After her SLCT diagnosis (2019), the patient was referred to a genetic counsellor and genetic testing. Routine and molecular karyotype performed were normal (46, XX). Whole exome sequencing (WES) revealed that she is heterozygous for a new frameshift variant of the DICER1 gene, in exon 16 (c.2685dupA), consisting of a nucleotide duplication (NM_001195573: c.2685dupA) that is responsible for preterm ending of DICER1 protein synthesis (p. Phe.896IIefs*5) [49]. This variant is characterized as pathogenic according to the criteria of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [50]. The method of sequencing was next-generation sequencing (NGS), and the variant was confirmed by multiple sequencing and “deep” reads (>100×). The timeline of our patient’s history is depicted in Figure 4.
2.6. Family History
Notably, the patient had a positive family history; namely, her maternal aunt had a history of rhabdomyosarcoma of the uterus, diagnosed at the age of 11 years and treated with chemotherapy, and subsequently she was in remission. She also had a history of MNG since the age of 14 years and had undergone total thyroidectomy at the age of 21 years, with no evidence of thyroid carcinoma. Her mother was found to carry the same pathogenic variant, but she was otherwise asymptomatic. To date, her mother refuses to complete the suggested diagnostic workup, and her aunt refuses to be tested for the variant.
3. Discussion
Herein, we present the case of a young Greek female patient with DICER1s, consisting of MNG and two sex-cord ovarian tumors, due to a novel pathogenic variant in the DICER1 gene.
DICER1s is characterized by a high predisposition for the development of a broad spectrum of benign and malignant neoplasms, expressed with a variety of signs and symptoms, depending on the physiopathological mechanisms involved [51]. MNG is a well-known component of the syndrome. Our patient has MNG with benign nodules, based on the ultrasonography and the FNA features, but she has been advised to regular follow-up, as thyroid carcinoma appears in 5–15% of cases [24]. While MNG itself does not necessarily indicate the presence of a DICER1 pathogenic variant, when combined with SLCT, it is highly suggestive of the syndrome [10]. SLCT is a very rare type of ovarian sex-cord stromal tumor; it nevertheless appears in great frequency among patients with DICER1s. Our patient experienced menstrual irregularities 2 years before the diagnosis and had hyperandrogenemia and suppressed serum FSH and LH levels, abnormalities that were reversed after treatment. The abdominal pain was attributed to mass effect and subsequent obstructive phenomena in the pelvis. Until now, 6 years later, our patient has no evidence of recurrence. JGCT is another type of ovarian sex-cord stromal tumor that has been rarely associated with germline DICER1 pathogenic variants. DICER1s is most associated with SLCTs, while JGCT has been associated mainly with somatic DICER1 mutations [52]. There have been two reports of probable germline-associated DICER1s JCGTs: a patient with DICER1-related disorders presented with JGCT at the age of 16 and another patient whose second-degree cousin had pleuropulmonary blastoma developed JGCT at the age of 2 [16]. Our patient presented with an asymptomatic JGCT as a second sex-cord stromal tumor of the ovary that turned out to be stage 1, and until now, she is free of recurrence.
Although most conditions related to DICER1s occur in infancy and childhood, age distribution seems to vary widely. Our patient was diagnosed with a DICER1 pathogenic variant at the age of 19 years, and her mother is still asymptomatic at the age of 50 years, despite the positive genetic testing. Haley et al. have described a case of a 7-year-old female with SLCT, while the mother has been asymptomatic and not tested for the mutation until she was diagnosed with SLCT at the age of 38 years [40]. Despite the autosomal dominant manner of inheritance, carrying a pathogenic variant of the DICER1 gene does not always result in the development of the syndrome, indicating its unknown penetrance. It is thought that by the ages of 10 and 50 years, approximately 5% and 19% of patients with a germline DICER1 pathogenic variant will develop a neoplasm, respectively, with females being at a significantly higher risk than men [53]. Many heterozygous individuals may remain asymptomatic until a second somatic mutation is acquired, involving a crucial codon that affects DICER1 activity [9,14]. In our case, the patient’s mother, even if she has positive genetic testing, is still asymptomatic, whereas the maternal aunt had rhabdomyosarcoma of the uterus and MNG at the ages of 11 and 14 years, respectively.
Genetic testing for DICER1 germline pathogenic variants must be offered in all patients with DICER1-related conditions, such as PPB, cystic nephroma, SLCT, cervical embryonal rhabdomyosarcoma, and pituitary and pineal blastoma, with or without positive family history [10,13]. Of course, all first-degree relatives should be screened for the patient’s specific DICER1 variant. Imaging surveillance recommendations by system have also been suggested, depending on the age and time of diagnosis [13]. When establishing the diagnosis, the significance of genetic counseling and clinical surveillance must be highlighted in order to early recognize and treat complications of the syndrome, not only in patients but also in family members.
To our knowledge, this is the first case associated with DICER1s, consisting of MGN and both types of sex-cord stromal tumors, due to the pathogenic variant in exon 16 c.2685dupA (Phe.896IIefs*5), which, up to date, is a novel pathogenic DICER1 variant expressed with SLCT, JGCT, and MNG in our patient. Interestingly, the mother is asymptomatic, suggesting the need for further research to elucidate the underlying mechanisms.
4. Conclusions
DICER1s is a rare clinical entity predisposing to the development of benign and malignant tumor and nontumorous conditions. Although it is inherited in an autosomal dominant manner, more data is needed to clarify the clinical fingerprint of DICER1 pathogenic variants, in order to explain symptomatic cases with common or uncommon components of the syndrome with asymptomatic, rather old mothers. Co-occurrence of MNG in childhood with other rare neoplasms, such as SLCT and cystic nephroma, strongly suggests the disorder. Prompt diagnosis is of clinical significance, regarding appropriate monitoring, early recognition and targeted treatment of tumors, identification of other family members and providing proper genetic counselling and testing. There are still unanswered questions regarding the parameters that determine whether particular DICER1s entities are malignant or undergo transformation. In the future, we should focus on developing targeted therapeutic approaches, refining screening protocols to accurately diagnose conditions associated with the syndrome, and improving the identification of at-risk individuals.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Schultz K.A.P. Harris A. Messinger Y. Sencer S. Baldinger S. Dehner L.P. Hill D.A. Ovarian Tumors Related to Intronic Mutations in DICER 1: A Report from the International Ovarian and Testicular Stromal Tumor Registry Fam. Cancer 20161510511010.1007/s 10689-015-9831-y 26289771 PMC 4699873 · doi ↗ · pubmed ↗
- 2Riascos M.C. Huynh A. Faquin W.C. NoséV. Expanding Our Knowledge of DICER 1 Gene Alterations and Their Role in Thyroid Diseases Cancers 20241634710.3390/cancers 1602034738254836 PMC 10814847 · doi ↗ · pubmed ↗
- 3Lee Y. Kim M. Han J. Yeom K.-H. Lee S. Baek S.H. Kim V.N. Micro RNA Genes Are Transcribed by RNA Polymerase IIEMBO J.2004234051406010.1038/sj.emboj.760038515372072 PMC 524334 · doi ↗ · pubmed ↗
- 4Han J. Lee Y. Yeom K.-H. Kim Y.-K. Jin H. Kim V.N. The Drosha-DGCR 8 Complex in Primary Micro RNA Processing Genes Dev.2004183016302710.1101/gad.126250415574589 PMC 535913 · doi ↗ · pubmed ↗
- 5Denli A.M. Tops B.B.J. Plasterk R.H.A. Ketting R.F. Hannon G.J. Processing of Primary Micro RN As by the Microprocessor Complex Nature 200443223123510.1038/nature 0304915531879 · doi ↗ · pubmed ↗
- 6Yi R. Qin Y. Macara I.G. Cullen B.R. Exportin-5 Mediates the Nuclear Export of Pre-Micro RN As and Short Hairpin RN As Genes Dev.2003173011301610.1101/gad.115880314681208 PMC 305252 · doi ↗ · pubmed ↗
- 7Bernstein E. Caudy A.A. Hammond S.M. Hannon G.J. Role for a Bidentate Ribonuclease in the Initiation Step of RNA Interference Nature 200140936336610.1038/3505311011201747 · doi ↗ · pubmed ↗
- 8Hammond S.M. Dicing and Slicing FEBS Lett.20055795822582910.1016/j.febslet.2005.08.07916214139 · doi ↗ · pubmed ↗