Whole Exome Sequencing in Drug-Induced Angioedema Caused by Angiotensin-Converting Enzyme Inhibitors: A Pilot Study in Five Patients
Alejandro Mendoza-Alvarez, Juan-Antonio Martinez-Tadeo, Eva Perez-Rodríguez, Javier Barrios-Recio, Jose-Carlos García-Robaina, Almudena Corrales, Itahisa Marcelino-Rodríguez, Jose-Miguel Lorenzo-Salazar, Rafaela González-Montelongo, Carlos Flores, Ariel Callero

TL;DR
This study explores genetic factors linked to angioedema caused by ACE inhibitors in five patients using whole exome sequencing.
Contribution
The study identifies potential genetic variants in F5 and ACE genes associated with drug-induced angioedema.
Findings
The rs6025 variant in the F5 gene was found in all patients, linked to increased blood clotting.
A common ACE gene variant (rs4343) was identified in two patients.
A novel ACE variant (rs142947404) was found in one patient, not previously studied in AE-ACEi.
Abstract
Background and Objectives: One of the most common causes of drug-induced angioedema (AE-DI) is related to reduced bradykinin breakdown after the use of certain medications. This is the case for forms of AE-DI due to the use of angiotensin-converting enzyme inhibitors (ACEi), which are used for the treatment of cardiovascular conditions. The causes of AE are not clear in these patients. Given the limited number of AE-ACEi genetic loci identified by genome-wide association studies, we opted to assess the utility of NGS of a panel of relevant genes to identify candidate genetic risk factors in severely affected patients. Methods: Five hypertensive patients from unrelated families with clinical AE-ACEi were included in the study. Whole-exome sequencing, variant calling, and annotation techniques were used. ANNOVAR v18.04.16 was used to annotate the variant calls. The resulting variants for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
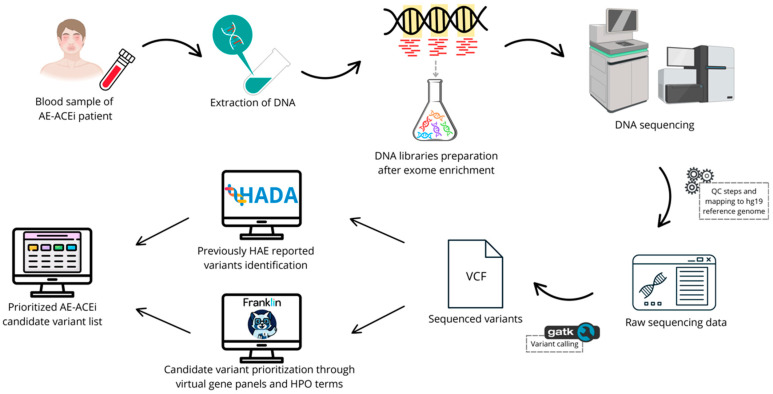 Figure 1
Figure 1| Gene | Individual ID | Total Depth (ref/alt) | HGVS Coding | HGVS | Effect | ACMG Class | ACMG Criteria | gnomAD | CADD Phred Score | MSC 95% HGMD | Predicted Effect [Reference] |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| AM2419 | 156 (0/156) | p.Gln534Arg | Missense | Likely | PM1 (moderate), PM5 (moderate), PM2 (supporting) | Absent | 0.255 | 12.286 | Increased blood clotting [ | |
| AM2450 | 124 (0/124) | ||||||||||
| AM2498 | 145 (75/70) | ||||||||||
| AM2569 | 121 (0/121) | ||||||||||
| AM2712 | 125 (0/125) | ||||||||||
|
| AM2419 | 101 (0/101) | none | Regulatory (intronic) | Benign | BA1 (stand-alone), BP4 (strong), BP6 (moderate) | 0.7967 | 0.446 | 11.653 | Higher risk of ACEi induced | |
| AM2450 | 117 (0/117) | ||||||||||
| AM2498 | 96 (1/95) | ||||||||||
| AM2569 | 87 (47/40) | ||||||||||
| AM2712 | 102 (0/102) | ||||||||||
|
| AM2498 | 213 (138/75) | p.Thr776= | Synonymous | Benign | BA1 (stand alone), BP6 (very strong), BP4 (strong), BP7 (supporting) | 0.4727 | 0.083 | 0.177 | Increased ACE | |
| AM2712 | 206 (136/70) | ||||||||||
|
| AM2450 | 88 (39/49) | p.Asn1036Lys | Missense | Uncertain | BP4 (strong), BP1 (supporting), PM2 (supporting) | 9.6 × 10−4 | 22.7 | 0.177 | Not reported in the scientific |
- —the Ministerio de Ciencia e Innovación
- —the European Regional Development Funds “A way of making Europe” from the European Union; Cabildo Insular de Tenerife
- —Instituto Tecnológico y de Energías Renovables (ITER)
- —Fundación Canaria Instituto de Investigación Sanitaria de Canarias
- —Spanish Society of Allergy and Clinical Immunology Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCoagulation, Bradykinin, Polyphosphates, and Angioedema · Peptidase Inhibition and Analysis · PI3K/AKT/mTOR signaling in cancer
1. Introduction
Drug-induced angioedema (AE-DI) is a medical condition often associated with reduced bradykinin (Bk) degradation following the use of specific medications [1]. A prominent example is AE-DI triggered by angiotensin-converting enzyme inhibitors (ACEi) used as a treatment for cardiovascular conditions, referred to as AE-ACEi. These medications interfere with the function of the angiotensin-converting enzyme, leading to an excessive accumulation of Bk in some patients [2]. AE-ACEi is uncommon, but it is calculated that the 12-month prevalence of AE-ACEi ranges from 0.004% to 0.026%. This percentage of affected patients varies between populations. Although the clinical manifestation of ACEi-AE is typically mild, there have been instances of fatal outcomes resulting from upper airway angioedema (AE) and subsequent obstruction [3]. Principal challenges in AE-ACEi patient management are related to differential diagnosis (AE-ACEi must be differentiated from other types of AE), diagnosis delay (influenced by the differential diagnosis), lack of specific treatment (management focuses primarily on discontinuation of ACEi intake), risk of lethal complications (mainly caused by upper airway obstruction), and genetic variability (ancestry and genetic factors influence in AE-ACEi susceptibility) [2].
Genetic testing in Bk-mediated AE (AE-BK) mostly focuses on rare hereditary forms, relying on conventional methods such as Sanger sequencing, multiplex ligation-dependent probe amplification (MLPA), and PCR. However, these genetic tests are rapidly evolving towards the use of next-generation sequencing (NGS), enabling highly efficient and holistic high-throughput genetic assessments. This transformation has paved the way for the identification of disease-causing variants, even in conditions with small genetic contributions. We have exposed the nuances of AE-ACEi, shedding light on the AE-DI forms, their connection to medication use, and the evolving methods for genetic testing, including whole exome sequencing (WES), as a powerful transformative tool to better understand and precisely manage this multifaceted condition in patients [2]. Given the limited number of AE-ACEi genetic loci identified by genome-wide association studies, we opted to assess the utility of NGS of a panel of relevant genes to identify candidate genetic risk factors in severely affected patients.
2. Materials and Methods
2.1. Patient Description
Five hypertensive patients from unrelated families with clinical AE-ACEi suspicion living in the Canary Islands (Spain) were recruited for this study (Table 1). Enalapril was the most frequently used drug among the patients in this study. They maintained treatment with ACEi for more than 15 months. The patients had at least one life-threatening episode of AE with dyspnea and oxygen saturation < 90%. The oropharynx was the most common location. All patients required assistance in the critical care unit. Antihistamines and corticosteroids were not effective. Complement studies were normal in all patients. The patients included in this study did not have any underlying pathology or chronic treatment that could influence the severity of AE-ACEi symptoms.
2.2. Sequencing, Identification, and Functional Annotation of Exome Variants
A total of 4 mL of peripheral blood was required for DNA extraction with Illustra^TM^ blood genomicPrep kit (GE Healthcare; Chicago, IL, USA). DNA concentration was assessed using the dsDNA BroadRange Assay Kit for the Qubit^®^ 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) (Figure 1). DNA sequencing libraries were prepared using the DNA Prep with Exome 2.0 Plus Enrichment Kit (Illumina, San Francisco, CA, USA), with fragment sizes and concentrations assessed on a TapeStation 4200 (Agilent Technologies, Santa Clara, CA, USA) and sequencing obtained with a NovaSeq 6000 Sequencing System (Illumina, San Francisco, CA, USA) with paired-end 101-base reads. PhiX was loaded and sequenced at 1% as an internal control for the experiments.
The resulting sequencing reads were preprocessed with bcl2fastq v2.18 and mapped according to the hg19/GRCh37 reference genome with Burrows-Wheeler Aligner v0.7.15 [4]. Secondly, BAM files were processed with Qualimap v2.2.1 [5], SAMtools v1.3 [6], BEDTools [7], and Picard v2.10.10 (http://broadinstitute.github.io/picard, accessed on 22 September 2020) for quality control steps. Variant calling of small germline variants was performed using the Genome Analysis Toolkit (GATK) v.3.8 for the detection of nucleotide substitutions (SNVs) and small indels (<50 bp) following best practices [8]. The pipeline description is publicly available (https://github.com/genomicsITER/benchmarking/tree/master/WES, accessed on 17 January 2024).
Candidate “PASS” variants were filtered using SAMtools and VCFtools, combined with more restrictive sequencing metrics such as depth of coverage per position (≥20×), genotype quality (≥100), and mapping quality (≥50).
Identification of true candidate genetic variants causing AE-ACEi requires in-depth filtering strategies, such as gene biological function, gene location, allele frequency in reference populations, and possible disease relations contemplated on ClinVar [9], and The Human Gene Mutation Database [10]. ANNOVAR software [11] was used to incorporate this information in the previously filtered step, in combination with the pathogenicity scores including the Combined Annotation-Dependent Depletion (CADD) in the context of the Mutation Significance Cutoff (MSC), among others. In order to maximize the identification of candidate variants responsible for symptoms, the pathogenic potential of the contemplated variants was extracted from InterVar software (January 2018 release), according to the American College of Medical Genetics and Genomics (ACMG) guidelines [12].
This bioinformatic process for previously sequenced variants and the annotation of required information for filtering steps were performed using the Teide-HPC Supercomputing facility (http://teidehpc.iter.es/en, accessed on 17 January 2024).
2.3. Prioritization of Potential Deleterious Variants
The Hereditary Angioedema (HAE) Database Annotation tool (HADA, http://hada.hpc.iter.es/, accesed on 17 April 2024) was used as the first prioritization strategy for previously annotated variants [13]. This tool allows ruling out undiagnosed hereditary angioedema (HAE) patients, as recommended by the International WAO/EAACI guideline for the management of affected patients in its last recommendations update [14]. HADA easily matches and provides previously published information in the related scientific literature for submitted variants, performing a rapid identification of variants affecting the protein function involved in this heritable subtype of angioedema.
We used the Franklin genomic platform for variant prioritization and clinical impact interpretation as a second tier for identifying genetic factors that could be responsible for AE-ACEi symptoms [15]. For this approach, we extracted from the scientific literature the most common affected genes in AE-ACEi and designed a virtual gene panel composed of ACE, BDKRB2, XPEPNP2, MME, F5, ETV6, DENND1B, and CRB1 [16]. This assessment was done by combining the search with a list of phenotypic abnormalities related to AE-BK (HP:0100665, HP:0025018, HP:0100540, HP:0002098, HP:0040315, HP:0031244, HP:0010783, HP:0002781, HP:0012027, HP:0011855).
2.4. Patient Population and Sequencing Summary
WES of the five patients provided an average of 6.58 Gb sequence, with an average of 100% of on-target reads and a median depth of 139.8×, and a transition/transversion ratio within the expected range (3.1 to 3.3).
3. Results and Discussion
AE is a relatively rare disease whose symptoms can be life-threatening. Due to the diverse etiology underlying symptom manifestation, it is key to identify the genetic risk factors using multiple strategies. In this sense, conducting a preliminary search in causal HAE genes is known to reduce the diagnostic odyssey of AE patients, as recommended by the International WAO/EAACI guideline (recommendation 3) [14]. The benefits of searching for variants in HAE causal genes have been proven by the identification of candidate variants with a protective effect against AE-ACEi [17]. In this first step, HADA did not reveal previously reported HAE causal variants present in recruited patients, reducing the possibility that the symptoms could be due to an undiagnosed HAE.
Secondly, sequencing data were also submitted to the Franklin platform to prioritize interesting likely deleterious variants responsible for inflammation symptoms (Table 2). Among the findings obtained by this tool, the variant c.1601C>T in the F5 gene was identified in all recruited samples, which has been previously associated with an increase in blood clotting in affected patients using exome data [18]. A genetic meta-analysis of AE-ACEi found the association of this variant of the factor V Leiden at a genome-wide significance level, reinforcing the evidence of its pathogenic effect [3].
Franklin also prioritized the intronic variant c.989-53 T>G in the CRB1 gene in all patients included in this study. This candidate variant has been associated with an increased risk of AE attacks because of ACEi intake in the ONTARGET dataset [16]. However, there is no further scientific information on this variant that provides additional evidence regarding the role of c.989-53 T>G in the pathogenic mechanism responsible for the symptoms.
The relevant function of the ACE gene in this disease makes the search for variants located in this gene a crucial step, allowing the identification of different candidate variants in the exome data. Moreover, ACE is one of the main components that inactivate the Bk activity. The synonymous c.2328 G>A genetic variant of ACE was found in two patients, AM_2498 and AM_2712. This variant was previously assessed to determine the association with AE-ACEi, specifically with captopril, and was associated with ACE activity [19]. In addition, the variant was reported as a possible risk factor for the development of AE-ACEi, likely due to its impact on the regulation of Bk levels and the ACE enzyme activity [20]. Although the c.2328 G>A in the ACE gene has been associated with AE-ACEi, the pathogenetic scores and population allele frequency of the variant support a benign classification. This scenario is common in clinical sequencing studies and highlights the intrinsic difficulties in the variant interpretation and its significance for the disease. The complexity lies in the fact that, by sequencing a larger number of genes, numerous variants are identified for which clinical relevance is uncertain, which complicates the prioritization, medical decision-making, and treatment prescription based on the data [21]. We also identified the ACE genetic variant c.3108 C>A in Patient AM_2450, which results in the change of asparagine to lysine (p.Asn1007Lys). This variant has not been assessed in AE-ACEi despite it being included in ClinVar as a variant of uncertain significance [22]. Some of the prediction scores such as SIFT and PROVEAN suggest a deleterious effect and its allele frequency in European populations is <0.1%. The findings on the ACE gene in this pilot study support the relevance of analyzing its variants to better understand the genetic predisposition to the disease.
In this study, significant limitations must be considered. The first constraint is the small sample size (five patients), which restricts the generalizability of the findings and limits the statistical power to establish strong associations between the identified genetic variants and ACEi-induced AE. A larger cohort will be necessary to confirm these preliminary observations and provide more robust conclusions. In this respect, this study should be considered a pilot study to evaluate alternative genetic screening strategies to identify novel genes governing AE-ACEi. Secondly, the study lacks functional data to validate the potential pathogenicity of the candidate variants. Without them, it is challenging to determine their biological impact and their role in the disease mechanism. This limitation also makes it difficult to exclude the involvement of certain variants and prioritize others that may emerge as stronger candidates when integrated with functional evidence. Future research incorporating larger patient cohorts and functional validation studies will be essential to refine the interpretation of genetic findings and enhance their clinical applicability. Among the strengths, it represents an innovative approach by using exome data to explore the genetic basis of AE-ACEi, expanding the identification of causal genetic variation and opening new research horizons beyond typical genetic screenings. Our findings are supported by evidence from the scientific literature and genetic databases, reinforcing the validity of the identified variants and their potential involvement in the pathogenesis of AE-ACEi. In addition, the use of advanced bioinformatics tools and variant prioritization strategies has allowed the identification of candidate genetic variants with previous evidence of association with AE-ACEi, suggesting that these results could be relevant for future studies with larger cohorts. Finally, our results also support the importance of further investigating variants in the ACE gene and other genes of the renin–angiotensin axis, which could contribute to the development of more precise strategies for the prevention and management of ACEI-induced AE.
In conclusion, we have explored the role of genetic variants in susceptibility to AE-ACEi by analyzing exome data from severely affected patients. Despite the limited sample size, we have uncovered the next steps for further genetic screening of AE-ACEi patients to improve the risk prediction and potentially prevent future episodes of AE induced by this drug.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Reshef A. Buttgereit T. Betschel S.D. Caballero T. Farkas H. Grumach A.S. Hide M. Jindal A.K. Longhurst H. Peter J. The Definition, Acronyms, Nomenclature, and Classification of Angioedema: AAAAI, ACAAI, ACARE, and APAACI DANCE Consensus J. Allergy Clin. Immunol.2024154398411.e 110.1016/j.jaci.2024.03.02438670233 · doi ↗ · pubmed ↗
- 2Marcelino I. Callero A. Mendoza-Alvarez A. Perez-Rodriguez E. Barrios-Recio J. Garcia-Robaina J.C. Flores C. Bradykinin-mediated angioedema: An update of the genetic causes and the impact of genomics Front. Genet.20191090010.3389/fgene.2019.0090031611908 PMC 6776636 · doi ↗ · pubmed ↗
- 3Mathey C.M. Maj C. Eriksson N. Krebs K. Westmeier J. David F.S. Koromina M. Scheer A.B. Szabo N. Wedi B. Meta-analysis of ACE inhibitor–induced angioedema identifies novel risk locus J. Allergy Clin. Immunol.20241531073108210.1016/j.jaci.2023.11.92138300190 · doi ↗ · pubmed ↗
- 4Li H. Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform Bioinformatics 2009251754176010.1093/bioinformatics/btp 32419451168 PMC 2705234 · doi ↗ · pubmed ↗
- 5García-Alcalde F. Okonechnikov K. Carbonell J. Cruz L.M. Götz S. Tarazona S. Dopazo J. Meyer T.F. Conesa A. Qualimap: Evaluating next-generation sequencing alignment data Bioinformatics 2012282678267910.1093/bioinformatics/bts 50322914218 · doi ↗ · pubmed ↗
- 6Li H. Handsaker B. Wysoker A. Fennell T. Ruan J. Homer N. The Sequence Alignment/Map format and SA Mtools Bioinformatics 2009252078207910.1093/bioinformatics/btp 35219505943 PMC 2723002 · doi ↗ · pubmed ↗
- 7Quinlan A.R. Hall I.M. BED Tools: A flexible suite of utilities for comparing genomic features Bioinformatics 20102684184210.1093/bioinformatics/btq 03320110278 PMC 2832824 · doi ↗ · pubmed ↗
- 8Mc Kenna A. Hanna M. Banks E. Sivachenko A. Cibulskis K. Kernytsky A. Garimella K. Altshuler D. Gabriel S. Daly M. The Genome Analysis Toolkit: A Map Reduce framework for analyzing next-generation DNA sequencing data Genome Res.2010201297130310.1101/gr.107524.11020644199 PMC 2928508 · doi ↗ · pubmed ↗