Desenmascarando al imitador: neumonía por citomegalovirus que inicia como CVID se diagnostica como deficiencia de GATA2 con dos mutaciones patológicas únicas
Andrés F Zea-Verano, Mónica Fernandes-Pineda

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
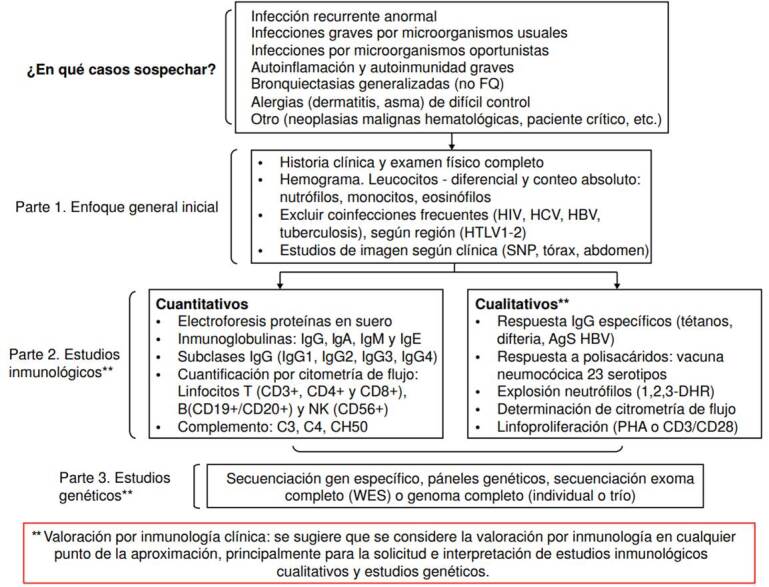 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmunodeficiency and Autoimmune Disorders · Cytomegalovirus and herpesvirus research · Blood disorders and treatments
A la pregunta 1. ¿Está de acuerdo con el diagnóstico inicial? ¿Cuáles serían los diagnósticos diferenciales?
Los diagnósticos diferenciales propuestos fueron:
- Error innato de la inmunidad, deficiencia predominantemente de anticuerpos:
b. ¿Inmunodeficiencia común variable?
c. ¿Agammaglobulinemia ligada al X, de aparición tardía?
d. ¿Defectos de la Inmunidad intrínseca o innata?
-
Neumonía recurrente secundaria a 1
-
Enfermedad diseminada por citomegalovirus, resuelta
-
Antecedente de neumonía no complicada por SARS-CoV-2
-
Infecciones crónicas y otras causas de inmunodeficiencia secundaria, descartadas
A la pregunta 2. ¿Qué estudios complementarios se solicitarían para este paciente?
Se consideró:
- Cuantificación de poblaciones linfocitarias extendidas; determinación de linfocitos T (CD4+ y CD8+), linfocitos B (CD19+/CD20+) y linfocitos NK (CD56)
- Subpoblaciones de linfocitos B para evaluar el cambio de isotipo y la presencia de linfocitos B de memoria
- Niveles valle de inmunoglobulina G (IgG)
- Secuenciación completa del exoma (WES) con énfasis en los genes relacionados con errores innatos de la inmunidad
Seguimiento y resultados de los estudios complementarios
El paciente ha recibido de forma continua sustitución con inmunoglobulinas por vía intravenosa, con buena tolerancia. Durante la evolución, por razones administrativas, las valoraciones por inmunología no le fueron autorizadas y no pudo asistir a los controles.
Las poblaciones linfocitarias extendidas y las subpoblaciones B no se determinaron. Los niveles valle de IgG fueron de 5,16 g/L en mayo del 2023, tras 12 meses de sustitución con inmunoglobulinas por vía intravenosa.
El exoma reportó dos variantes: c.938A>G (p.His313Arg) y c.937C>T (p.His313Tyr) en el gen GATA2. Se desconoce si estas variantes están en cis o en trans; ambas variantes fueron categorizadas como variantes de significado clínico incierto, de acuerdo con los criterios del American College of Medical Genetics - ACMG 1. Sin embargo, se debe recalcar que estas variantes nunca han sido reportadas en la literatura o en las bases de datos (Minor Allele Frequency, MAF < 0,000001) y la predicción bioinformática para ambas variantes fue de patógena o deletérea (SIFT, MutationTaster, MutPred, PolyPhen-2) con un CADD de 33 (Computer-Aided Drafting and Design, CADD) que nos habla de su patogenicidad. La variante p.His313Tyr ya ha sido identificada en la deficiencia de GATA2 como patológica y asociada con este error innato de la inmunidad 2.
El cuadro clínico, los hallazgos de laboratorio y el diagnóstico molecular, nos permitieron llegar al diagnóstico de deficiencia de GATA2. Este error innato de la inmunidad, también conocido como inmunodeficiencia 21 o MONOMAC, es una inmunodeficiencia primaria genética poco común, caracterizada disminución o ausencia de monocitos, células dendríticas circulantes y tisulares, linfocitos B y células asesinas, acompañada de propensión a infecciones por micobacterias, virus del papiloma y micosis, entre otras. Entre las manifestaciones clínicas más temidas, está el mayor riesgo de desarrollar neoplasias mieloides.
Conclusión
Dados los hallazgos de una médula ósea con aumento de células y monocitos ligeramente aumentados durante la hospitalización (560 células/ μl en mayo del 2022), se decidió valorar la evolución de los monocitos y se encontraron 900 células/μl en mayo del 2023. Por todo esto, se encuentra en seguimiento y vigilancia por el gran riesgo de transformación a una leucemia mielomonocítica crónica dado su diagnóstico genético. Actualmente, está siendo sometido a los estudios haplogenéticos pertinentes, para considerar un trasplante de médula ósea como opción curativa.
La discusión de la segunda parte de este artículo la centraremos en tres aspectos que consideramos claves, a saber: 1) enfoque del paciente adulto con sospecha de un error innato de la inmunidad; 2) errores innatos de la inmunidad más frecuentemente encontrados en adultos, y 3) deficiencia de GATA2.
Enfoque del adulto con sospecha de un error innato de la inmunidad
Los errores innatos de la inmunidad, tradicionalmente denominados inmunodeficiencias primarias, son un grupo heterogéneo de enfermedades monogénicas y fenocopias que afectan tanto a niños como a adultos 3. Las diferentes series estiman que entre el 30 y el 50 % de los casos de errores innatos de la inmunidad en todo el mundo, corresponden a personas mayores de edad 4.
En el enfoque del paciente con sospecha de un error innato de la inmunidad, la primera pregunta que nos debemos hacer es cuándo sospecharlo. A este respecto, algunas iniciativas -como la de la Jeffrey Modell Foundation- han trabajado en el establecimiento de los signos de alarma para un error innato de la inmunidad del adulto 5.
Los diez signos de alarma en el adulto son:
- Dos o más infecciones de oído nuevas en un año;
- Dos o más infecciones de senos paranasales en un año, en ausencia de alergia;
- Una neumonía por año por más de un año;
- Diarrea crónica con pérdida de peso;
- Infecciones virales recurrentes (resfriados, herpes, verrugas, condilomas);
- Necesidad recurrente de antibióticos endovenosos;
- Abscesos recurrentes de la piel o de los órganos internos;
- Moniliasis o infecciones micóticas de la piel o de otras localizaciones;
- Infecciones con micobacterias, y
- Historia familiar de un error innato de la inmunidad.
En nuestro medio, encontramos que los pacientes con un error innato de la inmunidad confirmado consultaron principalmente debido a infecciones recurrentes, un episodio de infección grave, infecciones oportunistas, síntomas de autoinflamación o autoinmunidad no claros o alteraciones en los resultados de laboratorio (hemograma) 6. No obstante, es importante destacar que hasta la tercera parte de los pacientes no cursaba con los signos clásicos de errores innatos de la inmunidad.
Teniendo en cuenta la literatura científica y la experiencia de más de 10 años del Servicio de Inmunología Clínica de la Universidad del Valle y el Hospital Universitario del Valle, proponemos un flujograma diagnóstico para los pacientes adultos con sospecha de un error innato de la inmunidad. Consideramos que, entre los signos de alarma de adultos, las bronquiectasias que no sean por fibrosis quística son un signo y una causa importantes de consulta en adultos 7 (figura 1), por lo cual esta debe tener una consideración especial adicional.
Figura 1Algoritmo de evaluación inicial del paciente adulto con sospecha de error innato de la inmunidad*AgS HBV: antígeno de superficie del virus de la hepatitis B; 1,2,3-DHR: dihidrorrodamina; FQ: fibrosis quística; HBV: virus de la hepatitis B; HCV: virus de la hepatitis C; HTLV1-2: virus linfotrópico de tipo 1 y 2 de las células T humanas; PHA: phytohemagglutinin; SPN: senos paranasales; HIV: virus de la inmunodeficiencia humana
Errores innatos de la inmunidad más frecuentemente encontrados en adultos
La literatura científica no está completamente unificada respecto al punto de corte para considerar adultos a los pacientes (15, 18, 21 o 25 años) con errores innatos de la inmunidad, y esta falta de uniformidad tiene su principal sustento en tres hechos:
- En muchos de los pacientes adultos con diagnóstico de un error innato de la inmunidad hecho durante la adultez, los síntomas se iniciaron en la niñez, lo cual significa, usualmente, un gran retraso diagnóstico 6;
- Los pacientes con diagnóstico de un error innato de la inmunidad hacen la transición a la edad adulta y pasan de los servicios de pediatría, donde fueron diagnosticados y tratados por años, a los servicios de medicina interna 8, y
- Están los pacientes cuyos síntomas y diagnóstico verdaderamente ocurren en la edad adulta.
De esta manera, sobre todo para los pacientes más jóvenes, es impreciso el momento en que se debe considerar como un error innato de la inmunidad de aparición en la edad adulta.
En forma global, podríamos afirmar que el grupo de las deficiencias predominantemente de anticuerpos -con la inmunodeficiencia común variable a la cabeza- representa cerca del 80 % de los pacientes adultos, diagnosticados en la edad adulta o en quienes se iniciaron síntomas pocos años antes de la adultez 9. Esto representa una gran ventaja, pero a la vez un gran reto. Si bien la mayoría de los pacientes con deficiencias de anticuerpos se pueden diagnosticar con estrategias relativamente sencillas en los estudios serológicos (determinación de inmunoglobulinas, subclases, anticuerpos específicos) y podrían beneficiarse de la sustitución de inmunoglobulinas, es también muy frecuente el no indagar más y quedarse satisfecho con un diagnóstico fenotípico sindrómico de hipogammaglobulinemia u otras deficiencias de anticuerpos, que puede estar enmascarando una etiología monogénica con consecuencias potencialmente más graves.
Los errores innatos de la inmunidad que clásicamente se presentan en la edad adulta son: el síndrome de Good, la linfopenia idiopática de CD4+, la deficiencia de GATA2, la inmunodeficiencia combinada de aparición tardía, la inmunodeficiencia común variable y la deficiencia selectiva de IgA 10, además de hipogammaglobulinemia de IgG o subclases en un grupo muy heterogéneo de sujetos. A pesar de ser estos los más frecuentes en la edad adulta, podemos afirmar que todos los grupos de errores innatos de la inmunidad presentes en la clasificación de la International Union of Immunological Societies (IUIS) pueden manifestarse por primera vez en la edad adulta 11.
Los mecanismos detrás del retraso en la aparición del cuadro clínico de un defecto que es congénito, ocurren por cambios epigenéticos, medioambientales, mutaciones hipomorfas y mutaciones somáticas 12; sin embargo, en la mayoría de los casos, no está claro por qué las manifestaciones clínicas se demoran tanto en el tiempo en presentarse.
El entendimiento de las bases moleculares de este grupo de errores innatos de la inmunidad nos ha permitido aprender que las mutaciones pueden producir efectos deletéreos parciales y dar origen a defectos hipomorfos que no se traducen en un cambio o en la pérdida completa de la función; esto, sumado a las mutaciones con aumento de la función -dominantes negativas, de penetrancia parcial, entre otras- hace que los posibles cuadros clínicos sean innumerables 13.
En la literatura científica hay varios artículos de revisión que presentan los errores innatos de la inmunidad de aparición en el adulto. No obstante, sugerimos dos, los cuales consideramos que presentan la información de una forma muy pedagógica: Primary immune deficiencies in the adult: A previously underrecognized common condition14 y Monogenic adult-onset inborn errors of immunity12.
Deficiencia de GATA2
La deficiencia de GATA2 fue recientemente descrita e integra diversas enfermedades y un amplio espectro clínico en un único diagnóstico genético. La anteriormente conocida como MonoMAC, o DCML (deficiencia de células dendríticas, monocitos, linfocitos B y células asesinas, entre otros), actualmente es categorizada dentro de los errores innatos de la inmunidad. Ocasionada por mutaciones heterocigotas en el gen GATA2, se manifiesta por la falta de un factor de transcripción clave de las células hematopoyéticas. Las alteraciones hematoinmunológicas se inician entre la segunda y la tercera década de la vida. Cursan con monocitopenia profunda, linfopenias y déficit de células B y células asesinas, junto con predisposición de los individuos al síndrome mielodisplásico y a la leucemia mieloide aguda 15.
El primer caso fue descrito en los Estados Unidos en 1989, y su diagnóstico genético se hizo después del 2011 16. Actualmente, la Organización Mundial de la Salud (OMS) la considera parte de los síndromes de predisposición al cáncer (específicamente neoplasias mieloides), hereditaria y poco frecuente. Se asocia con variantes patógenas de la línea germinal de GATA2 que pueden evolucionar a síndrome mielodisplásico o leucemia mieloide aguda, con potencial disfunción orgánica asociada 17.
Las manifestaciones clínicas tienen una considerable variabilidad, a menudo caracterizada por la propensión a las infecciones virales y fúngicas 18. Esto se evidencia por un patrón de infecciones recurrentes anormales, manifestadas por verrugas recurrentes causadas por el virus del papiloma humano (HPV), que son resistentes al tratamiento inicial; además, por infecciones fúngicas como aspergilosis, histoplasmosis y candidiasis, que se pueden desarrollar en forma muy grave o agresiva 15.
Entre otras manifestaciones sugestivas, se incluyen la sordera neurosensorial, la proteinosis alveolar pulmonar, las neoplasias hematológicas y la psoriasis, las cuales pueden iniciarse en la edad adulta y asociarse con hipogammaglobulinemia y con la reacción anormal de anticuerpos en los pacientes con infecciones recurrentes. Esto puede llevar a que se categorice inicialmente como una inmunodeficiencia variable común, sin pruebas genéticas 19^,^20.
El diagnóstico se enfoca desde el espectro inicial de manifestaciones clínicas 21. Los pacientes pueden presentar múltiples infecciones por virus, especialmente virus herpes (varicela zóster, virus de Epstein-Barr y citomegalovirus), agentes patógenos intracelulares y propensión a infecciones por micobacterias. Al igual que otros errores innatos de la inmunidad, se asocia con manifestaciones reumatológicas por desregulación inmunitaria (hiperlaxitud, osteoartritis, espondilitis anquilosante y artritis reumatoide seronegativa) 22.
En los cambios iniciales de los resultados de laboratorio, se encuentra neutropenia con monocitopenia grave 23. Sin embargo, en los casos de deficiencia de GATA2 que presentan monocitosis, se debe sospechar estadios de leucemia premielomonocítica 24.
La deficiencia de GATA2 se ha relacionado con neoplasias malignas hematológicas. Las mutaciones en la línea germinal de GATA2 son necesarias, más no suficientes, para desarrollar neoplasias mieloides, dado que no todos los pacientes con deficiencia de GATA2 evolucionan a neoplasias malignas hematológicas 23. El espectro del fenotipo de estos pacientes es amplio, y la penetrancia no está siempre dilucidada, lo que implica un reto diagnóstico 19. Sin embargo, las neoplasias malignas mieloides se presentan antes de los 40 años en el 80 % de los casos 25.
Al considerar los diagnósticos de exclusión para la deficiencia de GATA2, se deben tener en cuenta los siguientes diagnósticos diferenciales:
- anemia de Diamond-Blackfan, que se asocia con la anemia diseritropoyética y la trombocitopenia ligada al cromosoma X;
- infecciones virales recurrentes causadas por el virus del papiloma humano (HPV) con formación de verrugas;
- propensión a las infecciones por micobacterias, como el déficit de IL-12, y
- la menos frecuente agammaglobulinemia ligada al cromosoma X, que se diagnostica en la adultez y, generalmente, se asocia con manifestaciones clínicas leves o moderadas 25.
Respecto al tratamiento, el trasplante alogénico de médula ósea es el único tratamiento curativo para la deficiencia de GATA2, en el que se reversa el síndrome de insuficiencia medular 26. Sin embargo, dado el patrón de herencia autosómico dominante de esta enfermedad, se debe descartar el diagnóstico genético en los familiares al momento de considerar posibles donantes 27.
Conclusión
Los adultos pueden cursar con errores innatos de la inmunidad que pueden parecer simples deficiencias de anticuerpos y que, en realidad, están enmascarando una enfermedad genética mucho más grave. La hipogammaglobulinemia observada en el presente paciente, se asocia con las anomalías en las células plasmáticas identificadas en individuos con mutaciones de GATA2. Esta correlación destaca el papel crítico de GATA2 en sostener una población fuerte y saludable de células plasmáticas.
Nuestros hallazgos subrayan la imperiosa necesidad de practicar pruebas moleculares en los pacientes adultos, con el objetivo de excluir causas poco comunes como la deficiencia de GATA2, especialmente cuando hay anormalidades en las poblaciones linfocitarias y en los recuentos de monocitos.
Es importante ampliar nuestro panorama y aprovechar las herramientas moleculares, para llegar a una medicina de precisión, aun en la población adulta.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Richards S Aziz N Bale S Bick D Das S Gastier-Foster J Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Genet Med 20151740542410.1038/gim.2015.3025741868 PMC 4544753 · doi ↗ · pubmed ↗
- 2Donadieu J Lamant M Fieschi C de Fontbrune FS Caye A Ouachee M Natural history of GATA 2 deficiency in a survey of 79 French and Belgian patients Haematologica 20181031278128710.3324/haematol.2017.18190929724903 PMC 6068047 · doi ↗ · pubmed ↗
- 3Bousfiha A Moundir A Tangye SG Picard C Jeddane L Al-Herz W The 2022 update of IUIS phenotypical classification for human inborn errors of immunity J Clin Immunol 2022421508152010.1007/s 10875-022-01352-z 36198931 · doi ↗ · pubmed ↗
- 4Wang JJF Dhir A Hildebrand KJ Turvey SE Schellenberg R Chen LYC Inborn errors of immunity in adulthood Allergy Asthma Clin Immunol 2024206610.1186/s 13223-023-00862-838233962 PMC 10792788 · doi ↗ · pubmed ↗
- 5Modell V Orange JS Quinn J Modell F. Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes Immunol Res 20186636738010.1007/s 12026-018-8996-529744770 · doi ↗ · pubmed ↗
- 6Fernandes-Pineda M Matta-Cortés L Zea-Vera A. Errores innatos de la inmunidad Acta Médica Colombiana 20234910.36104/amc.2024.3092 · doi ↗
- 7Giraldo-Ocampo S Bonelo A Zea-Vera AF. B cell subsets in Colombian adults with predominantly antibody deficiencies, bronchiectasis or recurrent pneumonia Adv Respir Med 20229025426610.3390/arm 9004003536004955 PMC 9717342 · doi ↗ · pubmed ↗
- 8Cirillo E Giardino G Ricci S Moschese V Lougaris V Conti F Consensus of the Italian Primary Immunodeficiency Network on transition management from pediatric to adult care in patients affected with childhood-onset inborn errors of immunity J Allergy Clin Immunol 202014696798310.1016/jjaci.2020.08.01032827505 · doi ↗ · pubmed ↗