Characterizing the genetic diversity and population structure of Plasmodium knowlesi in Aceh Province, Indonesia
Pinkan P. Kariodimedjo, Nadia Fadila, Sydney R. Fine, Hidayat Trimarsanto, Chris Cotter, Leily Trianty, Michelle S. Hsiang, Jennifer Smith, Adam Bennett, Rintis Noviyanti, Farah N. Coutrier

TL;DR
This study examines the genetic diversity of Plasmodium knowlesi in Aceh, Indonesia, finding low diversity and distinct strains compared to Malaysia.
Contribution
The paper provides the first evidence of minimal genetic diversity and distinct P. knowlesi strains in Aceh, Indonesia.
Findings
P. knowlesi in Aceh showed low genetic diversity and low heterozygosity (HE = 0.63).
Strains from Aceh were genetically distinct from those in Malaysia based on clustering analyses.
Most infections were monoclonal, with a mean multiplicity of infection of 1.06.
Abstract
As in other parts of Southeast Asia, efforts to achieve or sustain malaria elimination in Indonesia have been threatened by the emergence of human infection with the primate species P. knowlesi. To understand the transmission dynamics of this species, investigation of P. knowlesi genetic diversity and population structure is needed. A molecular surveillance study was conducted in two phases between June 2014 and September 2018 at five primary health facilities in Aceh Province, Indonesia, an area nearing malaria elimination. Dried blood spot samples were collected from patients presenting with suspected malaria and testing positive for malaria by microscopy. PCR was performed for molecular confirmation and species identification. Forty-six samples were confirmed to be P. knowlesi, of which 41 were amplified with genotyping targeting ten known P. knowlesi microsatellite markers. For…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
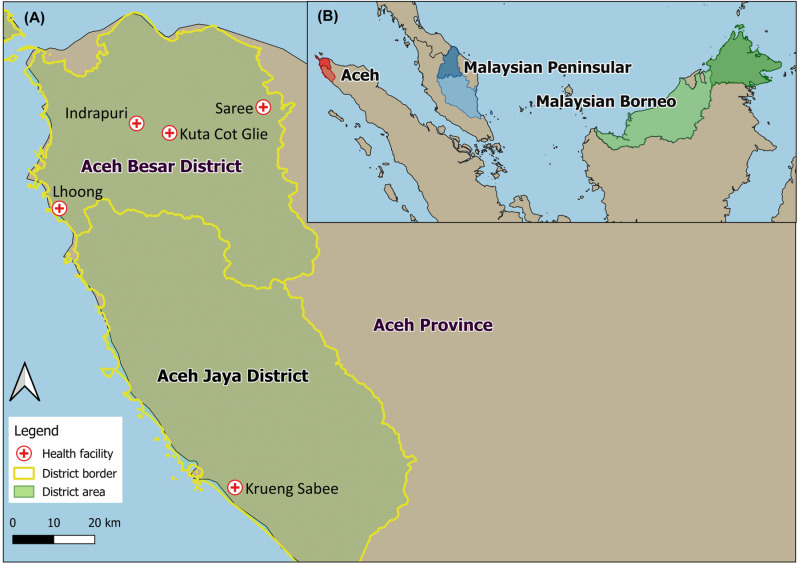 Fig 1
Fig 1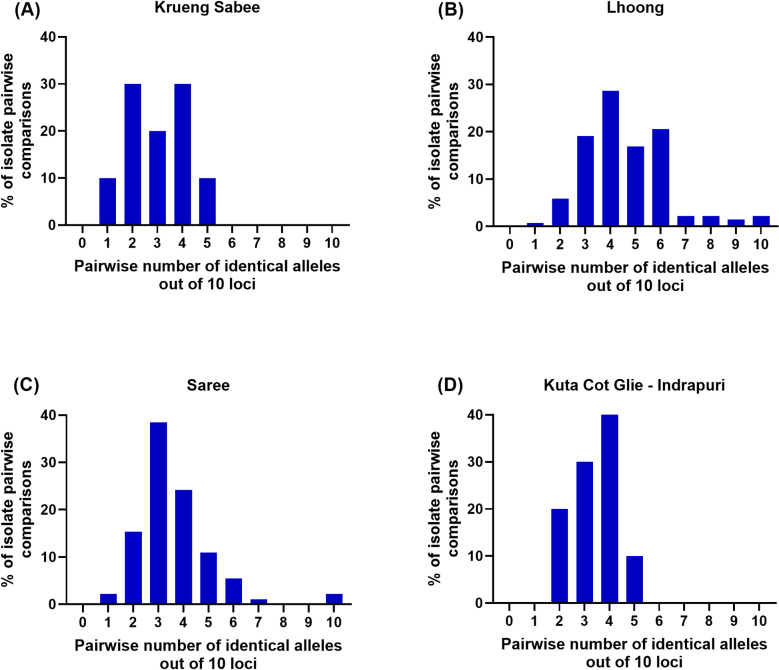 Fig 2
Fig 2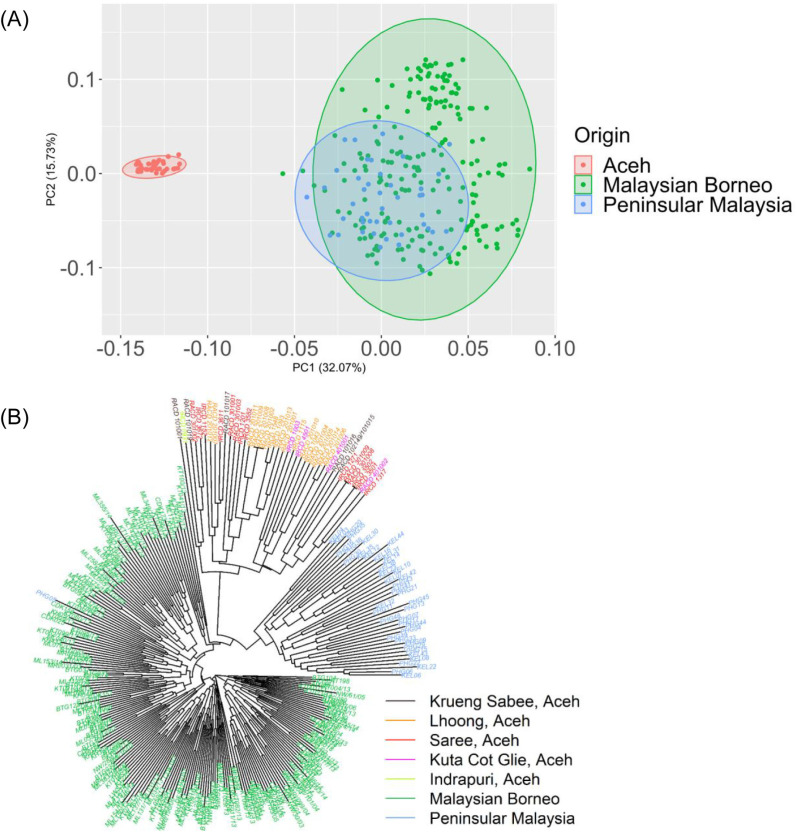 Fig 3
Fig 3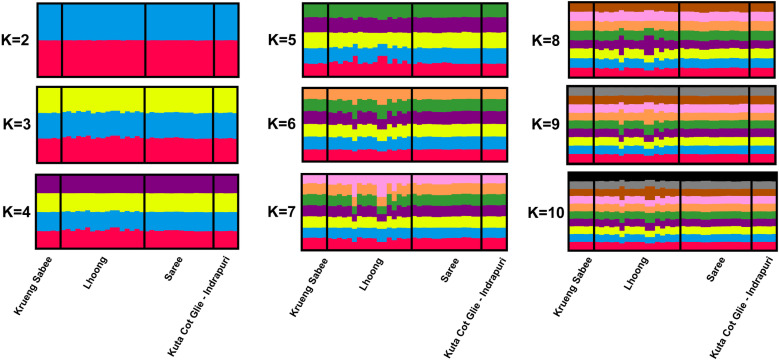 Fig 4
Fig 4- —http://dx.doi.org/10.13039/100000865Bill and Melinda Gates Foundation
- —http://dx.doi.org/10.13039/100000865Bill and Melinda Gates Foundation
- —http://dx.doi.org/10.13039/100000060National Institute of Allergy and Infectious Diseases
- —http://dx.doi.org/10.13039/100000865Bill and Melinda Gates Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMalaria Research and Control · Mosquito-borne diseases and control · Bird parasitology and diseases
Introduction
Similar to surrounding countries in Southeast Asia, malaria remains a critical public health concern in Indonesia. Indonesia experienced the second largest burden of malaria cases in the Southeast Asia Region in 2021, second only to India. Nevertheless, between 2015 and 2021, malaria incidence was reduced by 26% and the malaria mortality rate was reduced by 19% in Indonesia [1]. Malaria transmission in Indonesia is highly heterogenous, although areas in eastern Indonesia and Indonesian Borneo to the north have relatively intense transmission [2–4]. There are 372 provinces that have eliminated malaria, 83 that are considered low (<1 case/1000 population) transmission, and many others that have mid (1–5 cases/1000 population), high (>5 cases/1000 population), or unknown levels of transmission [5].
Historically, most malaria infections in Indonesia have been caused by Plasmodium falciparum or Plasmodium vivax [6,7]. However, a non-human primate species, Plasmodium knowlesi was recently detected in Indonesia. The first documented cases of P. knowlesi in Indonesia were identified in Sumatra, the western part of Indonesia, in 2014 [8–10]. Additionally, the mosquito vectors that carry P. knowlesi are common in the same region [11]. Macaca fascicularis and Macaca nemestrina, the long-tailed and pig-tailed monkeys, are the natural hosts of P. knowlesi and can withstand acute (natural and research-induced) infections and live normally with low-level parasitaemia [12,13]. These Macaca species inhabit almost all regions of Aceh Province [14–16]. The challenge in identifying this species earlier was largely due to errors in species identification by microscopy, and limited sensitivity, or cross-reactivity of species, with some molecular detection methods [17].
Understanding the genetic epidemiology of Plasmodium in an endemic area can inform control and elimination efforts [18,19]. Other groups have published data on the genetic diversity of P. falciparum [20] and P. vivax [20,21] in Indonesia. Evidence from these studies suggests that there is risk for distinct parasite subpopulations to persist within malaria-endemic communities, which may be below the limit of detection using standard diagnostics, such as microscopy and rapid diagnostic tests. Furthermore, forest workers and other migrant workers with patterns of mobility between endemic and non-endemic areas contribute to sustaining malaria transmission [22,23]. Particularly in areas of low transmission, understanding the genetic diversity and population structure of parasites can help to characterize transmission dynamics [24].
There is a limited understanding of the transmission dynamics of P. knowlesi, which has recently emerged as the dominant malaria species in Aceh Province, Indonesia. Increased awareness and knowledge of local patterns of malaria transmission and parasite relatedness can facilitate targeting of transmission reservoirs. To address this gap in evidence, this study aimed to characterize the genetic diversity and population structure of P. knowlesi using microsatellite genotyping.
Materials and methods
Study sites and sample collection
The study was conducted in Aceh Province, Indonesia, an area that has experienced a dramatic decline in malaria cases due to strengthened control and elimination efforts following the 2004 Indian ocean tsunami. The original data for this retrospective study were collected in two phases: Phase 1 (June 2014 to December 2015) [8,17] and Phase 2 surveillance studies (April 2017 – March 2018) [25]. The archive samples were accessed on 30 September 2019. The authors did not have access to information that could identify individual participants during or after data collection. Identifying details of each patient were removed by proper anonymization.
Patients who presented at a health facility with suspected malaria were invited to participate in the study. Written informed consent was obtained for venous blood collection before malaria treatment. Blood draw volume ranged between 250 uL-3 mL based on the age of the patient.
The overall Annual Parasite Incidence (API) in Indonesia between 2014–2018 was 0.55 [26]. In Phase 1, primary facilities with the highest number of malaria cases in Aceh Besar District were included, which represented 78% of all cases reported from the district in 2013. Although most districts in Aceh have reached elimination, in 2022, Aceh Jaya District had 32 cases with an API = 0.33 [5]. In Phase 2, a facility from neighbouring Aceh Jaya District was added due to its high burden P. knowlesi (Fig 1).
Maps of study sites and surrounding geographies.(A) Geographical locations of the five health facilities in this study (red plus sign). (B) Three geographic regions with established P. knowlesi transmission and with genetic data used in this study; Aceh (red), Peninsular Malaysia (blue), and Malaysian Borneo (green). (Source: Natural Earth, 2024).
Blood was collected to generate thick and thin blood smears for immediate diagnosis, to generate dried blood spots (DBS), and for collection in a heparin or EDTA tube for future analysis. Blood smears were fixed and stained with 3% Giemsa and read per national guidelines by a microscopist at the health facility. A thin smear was considered negative if no parasites were observed in 100 high-power fields. If parasites were identified, an additional 100 high-powered fields were examined to determine species. An expert level-certified microscopist performed quality assurance at the Banda Aceh Provincial Hospital for all positive malaria cases and 10% of randomly selected negatives. Results from the health facilities were confirmed by microscopists at the District and Provincial Health Offices.
DNA extraction and molecular identification
DNA extraction from the blood samples was performed at the Eijkman Institute, Jakarta, using the QIAamp DNA Mini Kit (Qiagen, Germany). Species identification for P. falciparum, P. vivax, P. malariae, and/or P. ovale was performed using nested PCR targeting the18S rRNA gene [27] and identification of P. knowlesi was performed using the PCR targeting ssrRNA [28]. PCR products were visualized by gel electrophoresis on 1.5% agarose gel. Positive P. knowlesi samples then underwent microsatellite genotyping.
Microsatellite genotyping of P. knowlesi
Microsatellite genotyping assays were conducted based on the ten trinucleotide markers (NC03_2, CD11_157, NC12_4, NC12_2, CD13_61, NC10_1, CD13_107, CD05_06, NC09_1) specific to P. knowlesi as previously described by Divis et al. [29]. Unique fluorescent dyes were assigned to each set of PCR primers to distinguish PCR products. Capillary electrophoresis was performed on an ABI 3100 Genetic Analyzer (Applied Biosystems, USA) using Genescan 600 LIZ as the molecular size standard (Applied Biosystems, USA). Genotyping analysis was conducted using GeneMapper Version 4.0 software (Applied Biosystems, USA) to identify genotypes. Electropherogram peaks were analyzed visually. To confirm genotyping quality, only allele peaks with a minimum height of 100 rfu were selected for analysis. The predominant allele plus any additional alleles with ≥ 33% of the height of the predominant allele were included in further analysis.
Data analysis
A polyclonal infection was defined as the presence of multiple alleles at one or more loci. Multiplicity of Infection (MOI) was defined as the maximum number of alleles at any given locus and is a proxy for the number of genotypes per infection. All the observed alleles were used to measure polyclonality and MOI; however, only the predominant allele—or alleles—at each locus was used for the population structure analysis. The expected heterozygosity (H_E_), including factors of population diversity, and fixation index (F_ST_), is a measure of the distance between populations and was calculated using Arlequin software version 3.5. Pairwise identical alleles of each isolate were determined using GenAlEx version 6.5 within Microsoft Excel. A website-based software, LIAN (http://guanine.evolbio.mpg.de/cgi-bin/lian/lian.cgi.pl), was used to define the index of association (I_A_^S^), which is a measure of multilocus linkage disequilibrium (LD). Neighbour-joining tree and principal coordinate analysis (PCoA) plots (genetic distance matrix) were constructed by the ape package in R version 3.6.3.
We conducted cluster identification by Bayesian analysis using STRUCTURE software (Pritchard Lab, Stanford University) with parameters set according to Divis et al. [29]. The most probable K-value was determined based on the highest ΔK value generated by Evanno’s method through STRUCTURE harvester (https://alumni.soe.ucsc.edu/~dearl/software/structureHarvester/). STRUCTURE harvester generated an output of cluster assignment indices for visualization of clusters through CLUMPP software version 1.1.2 (Rosenberg Lab, Stanford University) and Distruct software version 1.1 (Rosenberg Lab, Stanford University).
Ethical approval
Written informed consent was obtained from all adults or a parent/guardian for participants less than 18 years of age. This study was reviewed and approved by the Committee on Health Research Ethics of the National Institute of Health, Research and Development, Ministry of Health Republic of Indonesia (LB.02.01/5.2/KE.111/2014, LB.02.01/5.2/KE.211/2015, LB.02.01/5.2/KE.265/2016, LB.02.01/2/KE.083/2017), and the Committee on Human Research at the University of California, San Francisco (16-20220/171001, 16-20220/201201, 16-20220/232405, 16-20220/260683, 16-20220/289416, 13-10988/300323, 16-20220/337919). All the ethical approvals cover both the original studies and this retrospective study.
Results and discussion
Malaria speciation was conducted from which a total of 6 samples tested positive for P. falciparum and 29 samples tested positive for P. vivax. Forty-six samples were confirmed to be P. knowlesi, and 41 of these successfully amplified at all ten loci targeted by microsatellite genotyping (Table 1). Isolates were analyzed for marker properties, genetic diversity, and population structure. To facilitate group comparisons and due to their physical proximity, isolates from Kuta Cot Glie (n = 1) and Indrapuri (n = 1) were combined as a single group. With the exception of a few samples (n = 3) that presented multiple peaks, most samples had a single peak representing a single genotype (MOI = 1.064). Polyclonal infections were observed from Krueng Sabee (n = 1) and Lhoong (n = 2). The genetic diversity of the isolates obtained was considered moderate, with an overall H_E_ = 0.634. Krueng Sabee isolates exhibited the highest genetic diversity (H_E_ = 0.7).
Table 1: Genetic diversity of P. knowlesi infections collected from five primary health centers in Aceh.
Genetic diversity and structure
The diversity and genotyping for P. knowlesi microsatellite markers are summarised in S1 Table. Study isolates were characterized based on presence of a set of known P. knowlesi markers [29]. Based on the pairwise comparison analysis among genotypes, most isolates were identical by 3 or 4 markers (Fig 2). In samples from the Krueng Sabee and Saree regions, all ten loci had scores considered polymorphic. From the Lhoong and Kuta Cot Glie–Indrapuri regions, nine of ten polymorphic markers were observed; the tenth marker had the same allele size at locus CD13_107.
Pairwise comparisons of P. knowlesi isolates.These plots demonstrate the percentage of P. knowlesi isolate pairwise comparisons of 10 loci from (A) Krueng Sabee, (B) Lhoong, (C) Saree, and (D) Kuta Cot Glie–Indrapuri.
Multiple identical haplotypes were observed from the Lhoong and Saree regions: three identical haplotypes from the Lhoong population and two pairs of identical haplotypes in Saree. One isolate from Krueng Sabee scored two peaks at locus NC12_4. An isolate from Lhoong resulted in two peaks at marker CD08_61. Further, another isolate from Lhoong resulted in twin peaks at two markers, NC03_2 and NC10_1. The overall index of association amongst isolates from Aceh was IA^S^ = 0.008 (p-value = 1.5 × 10^−1^), which indicated low linkage disequilibrium (LD). All identical haplotypes were from the same population.
Overall population structure
PCoA and neighbour-joining tree analysis using the P. knowlesi sample population compared to P. knowlesi samples from Peninsular and Borneo Malaysia resulted in a distinct separation between groups of points (Fig 3). Overall, the results of the PCoA demonstrated a large scatter of points across the plot and no clear group of points (Fig 3a). Similarly, the results of the neighbour-joining tree analysis did not indicate a clear sub-population (Fig 3b). Based on the diagram, isolates from Aceh Province clustered closely together yet were distant from other known P. knowlesi populations. Additionally, isolates from Aceh Province clustered more proximally to isolates from Peninsular Malaysia compared to isolates from Malaysian Borneo.
Population structure of P. knowlesi from Aceh, Indonesia versus Malaysian Borneo and Peninsular Malaysia.(A) PCoA of P. knowlesi samples from Aceh (n = 41), Malaysia Borneo (n = 216), and Peninsular Malaysia (n = 43). (B) Unrooted neighbour-joining tree illustrating the genetic relatedness between P. knowlesi from Aceh, Malaysian Borneo, and Peninsular Malaysia.
Analysis using Evano’s test also suggested lack of substructure in the study population. The most probable number of clusters in this population was 2, given that the ΔK value was the highest (ΔK = 18.9) when K = 2 (S1 Fig, S2 Table). ΔK values were all < 1 when K ranged from 3–10. However, assessment of the differentiation between clusters 1 (K1) and 2 (K2) revealed no apparent local substructure in the Aceh Besar and Aceh Jaya populations (F_ST_ = 0.05202, P < 0.005) (Fig 4). Further, based on simulating values of K = 2–10, it appeared unlikely that there was substructure, as there was an equal admixture of both clusters (K1 and K2). It was concluded that there was only one true cluster in the population.
Population structure of P. knowlesi in Aceh.Bar plots illustrating the population structure at K = 2–10 in P. knowlesi isolates from Aceh. A single vertical bar represents an individual sample, and colors represent the K cluster (sub-population) defined by STRUCTURE. (K1 = red; K2 = blue; K3 = yellow; K4 = purple, K5 = green, K6 = orange, K7 = pink, K8 = brown, K9 = grey, K10 = black).
Discussion
In this genetic surveillance study of P. knowlesi conducted over a four-year period in Aceh Province, a near elimination setting, we analyzed 41 cases of P. knowlesi infection using microsatellite genotyping of 10 known markers. For samples within a site, nearly all or all loci were polymorphic. Across sites, multiple identical haplotypes were observed, though linkage distribution in the population was low. As reflected in HE, MOI, and F_ST_ values, the parasite population showed low diversity and low complexity. PCoA and neighbour-joining tree analyses provided evidence that P. knowlesi strains from Aceh were distinct from those reported in Malaysia, where the large majority of malaria infections over this period were P. knowlesi.
Genotyping of P. knowlesi with microsatellite markers has previously been performed on other Malaysian P. knowlesi populations [29,30]. To our knowledge, this is the first report of genetic diversity and population structure of P. knowlesi in Indonesia, specifically. Similar to P. knowlesi in Malaysia, we also observed a low index of association and minimal genetic differentiation amongst isolates. Each isolate was relatively geographically close to each other, without natural borders between regions. This phenomenon suggests possible inbreeding of the parasite and/or infrequent introduction of novel P. knowlesi genotypes into the population. Thus, a limited number of P. knowlesi genotypes spread with low genetic recombination occurrence. Low LD can be reinforced by recombination of P. knowlesi in mosquitoes following macaque blood meals containing multiple parasite genotypes [29]. Furthermore, we provide evidence that diversity of isolates was low, and most infections were caused by a single, predominant genotype of P. knowlesi with a low MOI number. Low MOIs from high and low endemic areas of P. knowlesi infections [30] in addition to populations without structure [31] have previously been reported.
We used the same primers to amplify ten microsatellite markers [29], allowing for comparison between the sites. Significant genetic isolation by geographic location was observed among clusters of isolates from Peninsular Malayasia, Malaysian Borneo, and Aceh Province. The findings show that population of different origins can be distinguished through this method.
There were some limitations of the study. First, while we provide characterization of the genetic diversity of P. knowlesi in this area, whole genome sequencing of the study isolates would provide a more robust dataset and the ability to compare P. knowlesi isolates to macaque isolates [32]. Second, our samples may not reflect the true population of samples in the study area. It is well established that RDTs and microscopy have limited sensitivity for detecting low-density infections. In passive surveillance of malaria among patients presenting with suspected malaria, low density infections may have been missed by microscopy. P. knowlesi has also been reported to cause asymptomatic infection [33] and our study did not include samples detected through active case detection. However, given the low diversity, complexity, and population structure of parasites across sites, it is unlikely that missed samples within sites would have altered these findings. Lastly, given the small number of samples, and limited geographic scope, we were not able to conduct analyses to examine trends over time, and findings cannot be generalized to other areas of the country or outside Indonesia.
Indonesia aims to eliminate the human malaria species by 2030 [34] and control P. knowlesi transmission to levels where it is not a significant burden. In 2021, Aceh Besar eliminated malaria, and Aceh Jaya maintained its low-endemicity status. Other progress towards reducing case loads and elimination overall has been made in terms of providing adequate access to malaria case management, strengthening malaria surveillance systems, and reenforcing community engagement and behaviour change communication programs [8,35]. Yet, locally acquired malaria cases continue to occur. Those who engage in forest-related work are at high risk for infection with P. knowlesi, as monkeys may be more present in the areas around logging sites and forest plantations [36,37]. The genetic data presented here is a positive indicator that correlates with the epidemiological data, suggesting that the overall low burden of P. vivax and P. falciparum, along with the low transmission of P. knowlesi, highlights that the Aceh region is moving closer to reaching the malaria elimination targets.
Conclusion
In a near-elimination setting in Indonesia, we found for the first time that P. knowlesi strains showed low diversity and low complexity, and were genetically distinct from Malaysian strains, suggesting highly localized transmission and limited connectivity to Malaysia. Ongoing genetic surveillance of P. knowlesi in Indonesia can inform tracking and planning of malaria control and elimination efforts.
Supporting information
S1 Fig ΔK value at each assumed K-value in STRUCTURE. The most probable cluster value is n = 2 based on the highest delta value from 20 replicates testing cluster value n = 2 – 10. Data must be used with population structure to test true admixture in sample group.(TIF)
S1 Table Allele scores of 10 microsatellite loci on P. knowlesi from Aceh, Indonesia. A summary of genotyping results of 41 P. knowlesi infections from Krueng Sabee, Lhoong, Saree, Kuta Cot Glie and Indrapuri. Multiple alleles are shaded in yellow and dominant alleles are shown in bold.(DOCX)
S2 Table ΔK value at each assumed *K-*value in STRUCTURE. The numeral data to graph delta K from sample group.(DOCX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World malaria report 2022 [Internet]. 2022 [cited 2023 Aug 1]. Available from: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022
- 2Sugiarto SR, Baird JK, Singh B, Elyazar I, Davis TME. The history and current epidemiology of malaria in Kalimantan, Indonesia. Malar J. 2022;21(1):327. doi: 10.1186/s 12936-022-04366-5 36372877 PMC 9661770 · doi ↗ · pubmed ↗
- 3Surjadjaja C, Surya A, Baird J. Epidemiology of Plasmodium vivax in Indonesia. Am J Trop Med Hyg. 2016;95(6 Suppl):121–32.27708185 10.4269/ajtmh.16-0093 PMC 5201218 · doi ↗ · pubmed ↗
- 4Elyazar IRF, Hay SI, Baird JK. Malaria distribution, prevalence, drug resistance and control in Indonesia. Adv Parasitol. 2011;74:41–175. doi: 10.1016/B 978-0-12-385897-9.00002-1 21295677 PMC 3075886 · doi ↗ · pubmed ↗
- 5Elyazar IRF, Gething PW, Patil AP, Rogayah H, Kusriastuti R, Wismarini DM, et al. Plasmodium falciparum malaria endemicity in Indonesia in 2010. P Lo S One. 2011;6(6):e 21315. doi: 10.1371/journal.pone.0021315 21738634 PMC 3126795 · doi ↗ · pubmed ↗
- 6Elyazar IRF, Gething PW, Patil AP, Rogayah H, Sariwati E, Palupi NW, et al. Plasmodium vivax malaria endemicity in Indonesia in 2010. P Lo S One. 2012;7(5):e 37325. doi: 10.1371/journal.pone.0037325 22615978 PMC 3355104 · doi ↗ · pubmed ↗
- 7Herdiana H, Cotter C, Coutrier FN, Zarlinda I, Zelman BW, Tirta YK, et al. Malaria risk factor assessment using active and passive surveillance data from Aceh Besar, Indonesia, a low endemic, malaria elimination setting with Plasmodium knowlesi, Plasmodium vivax, and Plasmodium falciparum. Malar J. 2016;15(1):468. doi: 10.1186/s 12936-016-1523-z 27619000 PMC 5020529 · doi ↗ · pubmed ↗
- 8Herdiana H, Irnawati I, Coutrier F, Munthe A, Mardiati M, Yuniarti T. Two clusters of Plasmodium knowlesi cases in a malaria elimination area, Sabang Municipality, Aceh, Indonesia. Malar J. 2018;17(1):186. doi: 10.1186/s 12936-018-2270-529720188 PMC 5932826 · doi ↗ · pubmed ↗