Coding-complete genome sequence of a GI-13 infectious bronchitis virus from commercial chicken in India
Henry M. Kariithi, Jeremy D. Volkening, Claudio L. Afonso, Mohamed Helmy, Pushparaj P. Chaudhari, Eduardo L. Decanini

TL;DR
This paper reports the complete genome sequence of a GI-13 infectious bronchitis virus strain from a chicken in India.
Contribution
The study provides the first complete genome sequence of a recombinant GI-13 IBV strain from India.
Findings
The virus was identified from a commercial broiler chicken using next-generation sequencing.
The strain is classified as GI-13, a known pathogenic type of infectious bronchitis virus.
Abstract
Infectious bronchitis virus (IBV) causes a highly contagious, acute upper respiratory disease in chickens characterized by nasal discharge, coughing, and rales. Here, the complete genome sequence of a recombinant GI-13 IBV strain ck/IN/A2332039-001/24 was sequenced from a choanal sample of a commercial broiler chicken in India using nontargeted next-generation sequencing.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
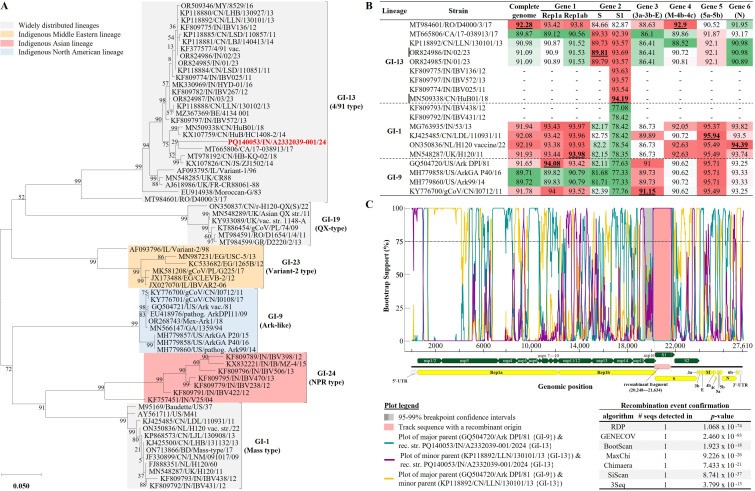 Fig 1
Fig 1| Gene | Consensus sequence | Variant sequence | Coverage depth | Variant frequency | Coding effect | |||
|---|---|---|---|---|---|---|---|---|
| Position | Codon | Amino acid residue | Codon | Amino acid residue | ||||
| Rep1ab (nsp1/2 [ZF-MF]) | 13,897 | CTT | Leu | Phe | 129 | 24% | SNP (nonsynonymous substitution) | |
| Rep1ab (nsp11/12 [RdRp]) | 14,298 | GCC | Ala | GC | Cys | 170 | 18.80% | SNP (nonsynonymous substitution) |
| Rep1ab (nsp13 [Hel]) | 15,153 | GTA | Tyr | GT | Val | 91 | 13.20% | SNP (nonsynonymous substitution) |
| Rep1ab (nsp14 [ExoN]) | 17,619 | TAT | Tyr | TA | Val | 81 | 37% | SNP (nonsynonymous substitution) |
| Spike (S) | 262 | TCT | Ser | Pro | 170 | 15.90% | SNP (nonsynonymous substitution) | |
| 356 | CAA | Gln | C | Pro | 92 | 19.60% | SNP (nonsynonymous substitution) | |
| 863 | GAC | Asp | G | Gly | 76 | 18.40% | SNP (nonsynonymous substitution) | |
| 3a | 83―85 | GGT-A | Val | G | - | 104 | 17.30% | Indel (disruptive inframe deletion of 3 nt) |
| 6b | 181 | GTG | Val | Met | 332 | 21.10% | SNP (nonsynonymous substitution) | |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAnimal Virus Infections Studies · Plant Virus Research Studies · Viral gastroenteritis research and epidemiology
ANNOUNCEMENT
Direct-nontargeted metagenomic next-generation sequencing, which simultaneously identifies and genotypes complex pathogens (1), frequently identified infectious bronchitis virus (IBV; family Coronaviridae [2]) during a project on epidemiological mapping of pathogens and enhancing biosecurity in commercial chickens in India, Middle-East, Turkey, and Africa (IMETA) region. The globally important IBVs comprise of 36 lineages within eight genotypes (GI―GVIII) and unclassified inter-lineage variants (3). We report here a complete genome sequence of IBV from West Bengal, India.
Choanal swabs from 50 13-day-old asymptomatic broilers were pooled in 2-mL Kylt Swab buffer and spotted on AniCard (NGS Sampling Kit; SAN Group Biotech GmbH; Emstek, Germany). Total RNA was extracted using Kylt RNA/DNA Purification kit followed by proprietary in-house removal of host-specific DNAs and paired-end sequencing (2 × 150 bp; Illumina MiSeq 300-cycle Reagent Kit v2) at SAN Group Biotech. Using default parameters, raw reads (n = 458,051) were adapter-/quality-trimmed using Trim Galore v1.6.10 (-q 8), host-filtered using BBTools bbduk 39.01, assembled using MEGAHITv1.2.9 (4), and variants/alleles frequency analyses (> 10% and SB <30) using LoFreq v 2.1.5 and bcftools v 1.19. The de novo-assembled consensus sequences were annotated using Geneious Prime v2024.0.7 (5) and aligned with published IBV sequences using MAFFT v7.511 (6) for phylogenetics (7). Recombination events were analyzed using RDP4 v 4.101 (8).
The identified 10,667 IBV-specific read pairs (BLASTn-based) were de novo-assembled into a consensus genome sequence (one contiguous contig; median coverage depth of 116×) of 27,599 nucleotide (nt) in length (excluding poly-(A) tail) with a 38.2% GC content. Open reading frames of this strain (ck/IN/A2332039-001/24) have typical avian coronavirus organization (5′UTR-[Rep1ab-S-3a/3b-E-M-4a/4c-5a/5b-N-6b]−3′UTR), including conserved features of structural proteins (S, E, M, and N), cleavage sites and lengths of nsp2-16, the primary S1/S2 cleavage motif (amino acid residues ^536^RLRR↓S ^540^), and N-linked glycosylation sites (n = 27) of its 3,495-nt-long S gene (2, 9). Table 1 shows polymorphisms present in the coding regions of Rep1ab, S, 3a, and 6b genes.
The complete genome of ck/IN/A2332039-001/24 is most similar to a Romanian GI-13 (4/91-type) strain D4000/3/17 (92.3% nt identity) but phylogenetically distinct from other Indian 4/91 viruses (Fig. 1A and B). Recombinant nature of the Indian strain was suggested by highest similarities of its spike region to 4/91 viruses and genome backbone (nonspike region) to GI-1 (Mass-type) and GI-9 (Ark-type) viruses (Fig. 1B). This was confirmed (statistically supported by seven RDP4 algorithms) by detection of a 1,387-bp-long recombinant fragment in its S1 sequence (Fig. 1C). An Ark-type is the closest relative of the major parent (strain closest to the sequence surrounding the recombinant region; 92.6% nt identity), and 4/91-type the minor parent (strain closest to the recombinant fragment; 96.8% nt identity) of the Indian strain. Use of Mass- and 4/91-type and absence of Ark-type vaccines in India suggest strain ck/IN/A2332039-001/24 recombined and eventually diverged from its parental strains long before its detection. Although not supported by variant frequencies (Table 1), the possibility of a mixed IBV infection cannot be ruled out.
(A) Phylogenetic relationship of the IBV GI-13 strain ck/IN/A2332039-001/24 identified in this study (highlighted in bold red font) with a selection of other IBVs based on full-length S1 nt sequences. The tree was constructed using maximum likelihood method and GTR model (MEGA v 11.0.13; 1,000 replicates) involving 68 sequences and 1601 positions. (B) Similarities (% nt identities) of genome sequence and gene coding regions (CDS) of the six IBV genes of strain ck/IN/A2332039-001/24 identified in this study with GI-1, GI-9, and GI-13 viruses. The color gradient code (from red-white-green) indicate highest (dark red) to lowest (green) nt identities for each of the genes. bold underlined numbers and dash (“-”) indicate highest identities and unavailable sequences for each CDS, respectively. (C) Recombination event identified and confirmed by seven out of nine RDP4 algorithms (bottom right Table) (8) in the S1 sequence of strain ck/IN/A2332039-001/24 with a GI-9 strain GQ504720/Ark DPI/81 and GI-13 strain KP118892/LLN/130101/13 as the predicted major and minor parents, respectively. The recombination event has breakpoints beginning 74 nt upstream of the S gene (genomic position 20,248) and ending at nt position 21,634, producing a recombinant fragment of 1,387 bp in length, which covers 79.3% (1,283/1,617 nt) of the S1 gene. Sequence names include GenBank accession numbers, two-letter abbreviated country of origin, isolate name, and year of sample collection/identification.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Afonso CL, Afonso AM. 2023. Next-generation sequencing for the detection of microbial agents in avian clinical samples. Vet Sci 10:690. doi:10.3390/vetsci 1012069038133241 PMC 10747646 · doi ↗ · pubmed ↗
- 2Woo PCY, de Groot RJ, Haagmans B, Lau SKP, Neuman BW, Perlman S, Sola I, van der Hoek L, Wong ACP, Yeh S-H. 2023. ICTV virus taxonomy profile: Coronaviridae 2023. J Gen Virol 104:001843. doi:10.1099/jgv.0.001843 PMC 1213507437097842 · doi ↗ · pubmed ↗
- 3Valastro V, Holmes EC, Britton P, Fusaro A, Jackwood MW, Cattoli G, Monne I. 2016. S 1 gene-based phylogeny of infectious bronchitis virus: an attempt to harmonize virus classification. Infect Genet Evol 39:349–364. doi:10.1016/j.meegid.2016.02.01526883378 PMC 7172980 · doi ↗ · pubmed ↗
- 4Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. doi:10.1093/bioinformatics/btv 03325609793 · doi ↗ · pubmed ↗
- 5Kariithi HM, Volkening JD, Leyson CM, Afonso CL, Christy N, Decanini EL, Lemiere S, Suarez DL. 2022. Genome sequence variations of infectious bronchitis virus serotypes from commercial chickens in Mexico. Front Vet Sci 9:931272. doi:10.3389/fvets.2022.93127235903135 PMC 9315362 · doi ↗ · pubmed ↗
- 6Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi:10.1093/molbev/mst 01023329690 PMC 3603318 · doi ↗ · pubmed ↗
- 7Tamura K, Stecher G, Kumar S. 2021. MEGA 11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027. doi:10.1093/molbev/msab 12033892491 PMC 8233496 · doi ↗ · pubmed ↗
- 8Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. 2015. RDP 4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev 003. doi:10.1093/ve/vev 00327774277 PMC 5014473 · doi ↗ · pubmed ↗