Stereoselective Synthesis of (R)-all-trans-13,14-Dihydroretinol and -Retinoic Acid
Paul Wienecke, Adriaan J. Minnaard

TL;DR
Scientists developed a new way to make specific vitamin A metabolites, which could help study their roles in the body.
Contribution
The paper introduces a stereoselective synthesis method for two vitamin A metabolites using specific chemical reactions.
Findings
A stereoselective synthesis of (R)-all-trans-13,14-dihydroretinol was successfully achieved.
The synthesis of (R)-all-trans-13,14-dihydroretinoic acid was also accomplished using key catalytic reactions.
The method uses E-selective HWE olefination and Ru(II)-catalyzed hydrosilylation as critical steps.
Abstract
Vitamin A (or all-trans-retinol) metabolites are involved in a wide range of cellular processes. However, the investigation of their biological role is hampered due to their very limited availability. Herein we report a stereoselective total synthesis of the vitamin A metabolites (R)-all-trans-13,14-dihydroretinol and -retinoic acid, applying an E-selective HWE olefination and a Ru(II) catalyzed intramolecular 7-endo-dig hydrosilylation as the key steps.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
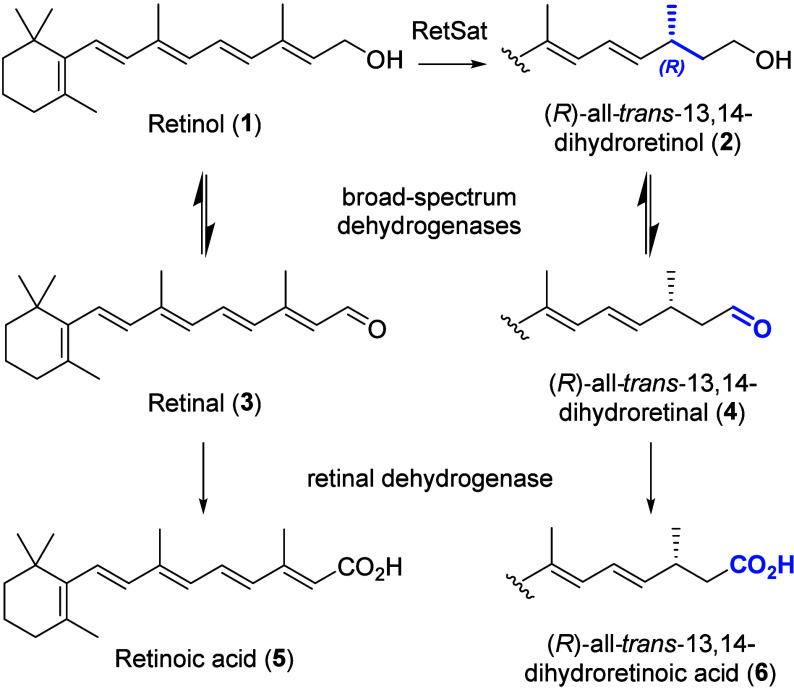 Figure 1
Figure 1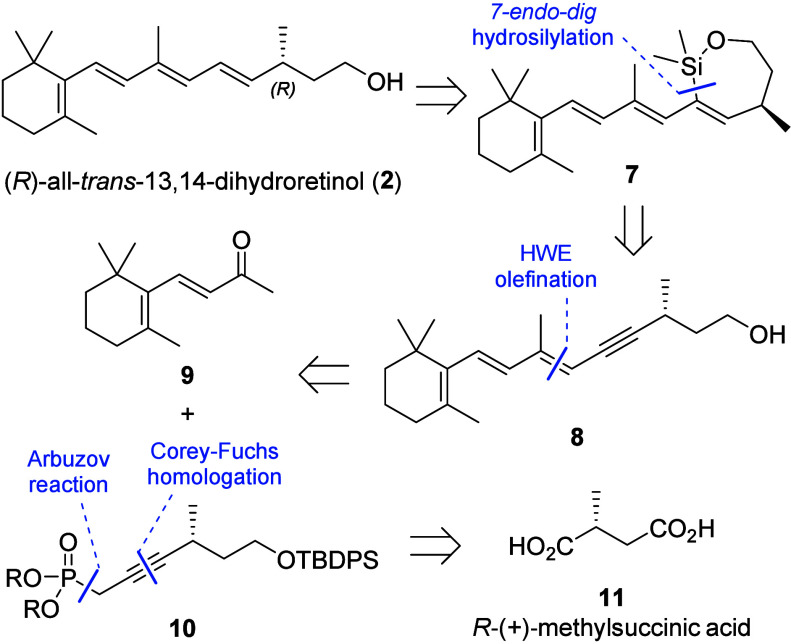 Figure 2
Figure 2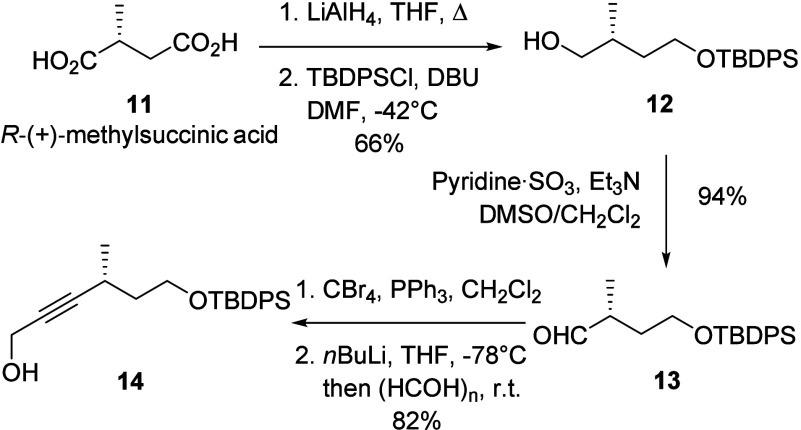 Figure 3
Figure 3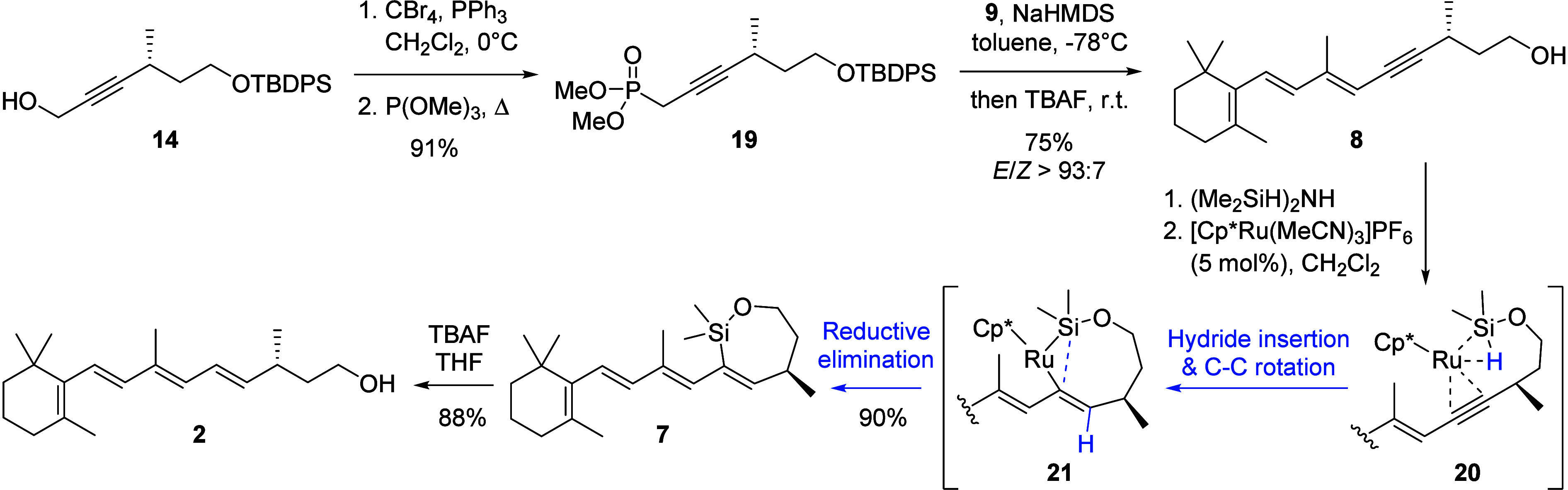 Figure 4
Figure 4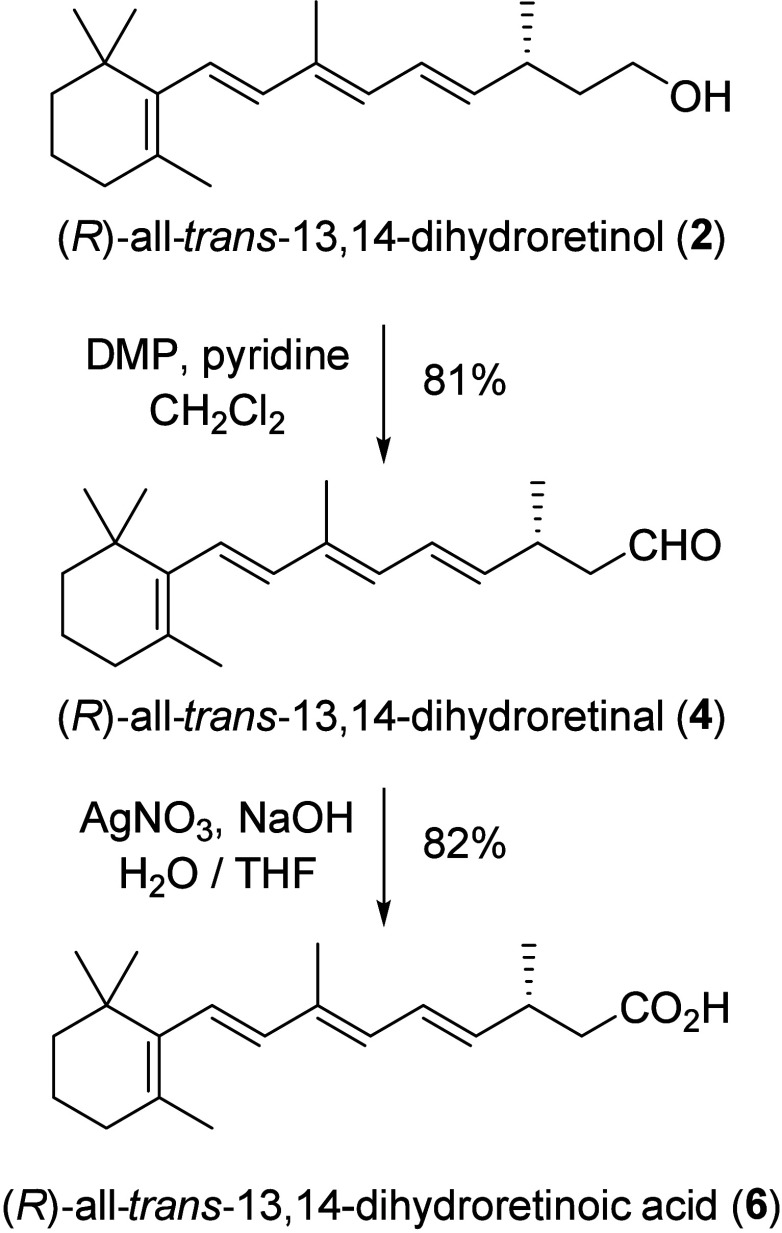 Figure 5
Figure 5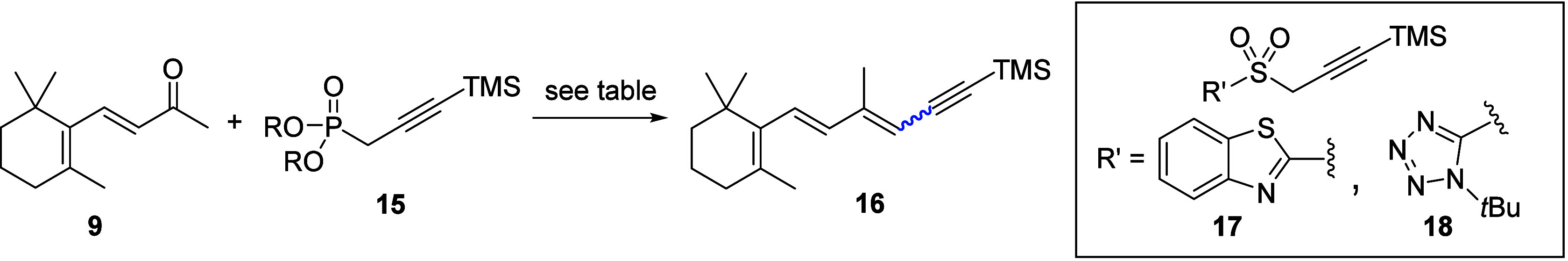 Figure 6
Figure 6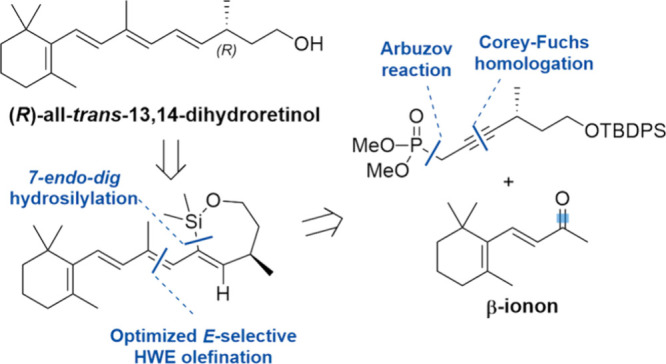 Figure 7
Figure 7- —National Institute of Allergy and Infectious Diseases10.13039/100000060
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNatural product bioactivities and synthesis · Traditional and Medicinal Uses of Annonaceae · Bioactive natural compounds
All-trans-retinol (1, Scheme 1), or vitamin A, is a fat-soluble, essential nutrient that humans need to obtain from their plant- or animal-based diet.^1^ In a broader sense, the term vitamin A also includes structurally related compounds, for instance, retinol metabolites and retinyl esters. This “vitamin A” plays important roles in a wide range of physiological processes, such as vision, cell differentiation, immune response, central nervous system development and embryogenesis.^2^ However, the very limited access to vitamin A metabolites hampers their investigation, leading to their poorly understood intertwinement with human body functions.^3^ A common metabolic pathway of all-trans-retinol (1) in vertebrates is the net hydrogenation to (R)-all-trans-13,14-dihydroretinol (2, Scheme 1) by the oxidoreductase retinol saturase (RetSat).^4−6^ This enzyme’s known involvement in adipocyte differentiation, liver metabolism, macrophage function and reactive oxygen species formation as well as its links to diabetes and tumor development underlines its central physiological role, but deeper understanding is lacking.^7^ The metabolic fate of (R)-all-trans-13,14-dihydroretinol (2) is similar to that of all-trans-retinol (1) and depends on the same enzymes (Scheme 1): First, 2 is oxidized to (R)-all-trans-13,14-dihydroretinal (4) by broad-spectrum dehydrogenases, followed by irreversible oxidation to (R)-all-trans-13,14-dihydroretinoic acid (6) by a retinal dehydrogenase.^8^
(R)-all-trans-13,14-dihydroretinol (2) and its corresponding carboxylic acid 6 are potent and selective activators of the retinoic acid receptor in vitro, likewise as their all-trans-retinol counterparts 1 and 5.^8^ However, the potency of 2 and 6 declines tremendously in vivo, probably below a physiological relevant threshold. Although they are metabolically more stable, a less efficient protein-mediated nuclear transport seems to account for this phenomenon.^9^ To date, possible extranuclear targets have not been investigated and the biological functions of (R)-all-trans-13,14-dihydroretinol (2) and its metabolites remain cryptic.^10^ The restricted access to these compounds poses a serious bottleneck for activity studies, for whom substantial amounts are needed. The single published synthesis allowed access and enabled the assignment of the absolute configuration,^6^ but the requirement for step-intensive sequences, such as diastereoselective formation of the stereocenter, and high excesses of CrCl_2_ and TlOH limits its applicability.
We aimed at a stereoselective total synthesis of (R)-all-trans-13,14-dihydroretinol (2) and the corresponding acid 6 to facilitate the elucidation of their functions in the human body. The double bond configurations in retinol and its derivatives play an important role in their bioactivities.^3^ Therefore, in planning the synthesis, we emphasized on eminently trans-selective alkene formations. Furthermore, the sensitivity of the conjugated alkenes toward oxidants, acids, heat and light had to be taken into account. In our retrosynthetic analysis, we envisioned (R)-all-trans-13,14-dihydroretinol (2) to be obtained from a stereoretentive desilylation after an alcohol-directed, intramolecular hydrosilylation of trien-yne 8 (Scheme 1). 7-endo-dig ring-closure to oxasilacycloheptene 7 would lead to an E-specific formation of the 11,12-double bond. In contrast, 6-exo-dig cyclization would also give the undesired *Z-*isomer.^11^ Considering the successful application of stereoselective olefinations in the synthesis of vitamin A derivatives,^6,12,13^ trien-yne 8 was planned to be accessed by an E-selective Horner-Wadsworth-Emmons (HWE) reaction of propargylic phosphonate 10 with β-ionone (9). We chose to synthesize phosphonate 10 from commercially available enantiopure R-(+)-methylsuccinic acid (11, the S-enantiomer is available as well), featuring the natural absolute configuration, via an Arbuzov reaction and a Corey-Fuchs homologation.
The synthesis of phosphonate 10 began with the reduction of (R)-(+)-methylsuccinic acid (11) to the diol by LiAlH_4_ (Scheme 2). Subsequent low-temperature TBDPS-silylation^14^ furnished the monoprotected diol 12 (Scheme 3) with good site-selectivity. Subsequent Parikh–Doering oxidation of the hydroxyl group gave aldehyde 13 in a very good yield. Following a Corey-Fuchs homologation approach, aldehyde 13 was first transformed into the corresponding dibromoalkene. Treatment with nBuLi and trapping the resulting alkynyllithium intermediate with paraformaldehyde, yielded efficiently propargyl alcohol 14 as the precursor for phosphonate 10.
In literature, HWE olefinations of β-ionone with propargylic phosphonates led to considerable amounts of Z-alkene byproducts.^15−17^ To improve E-selectivity of the envisioned olefination, we performed model optimization studies with readily available TMS-protected propargyl phosphonate 15, giving trien-yne 16 in good to very good yields and short reaction times (Table 1). It is worth mentioning that the Julia-Kocienski olefination reagents 17 and 18 did not give any conversion. As Julia-Kocienski olefinations of β-ionone (9) with nonstabilized aliphatic α-sulfonyl anions are known,^18^ it is likely that the stabilized propargylic anions exhibit insufficient reactivity for this transformation. An initial screening of bases for the deprotonation of the phosphonate (entries 1–4) revealed that NaHMDS gave the highest E-selectivity in comparison to the corresponding Li and K bases. Employing nBuLi instead of LiHMDS did not have a significant impact on the selectivity. Weaker bases such as NaOCH(CF_3_)2, Ba(OH)2, DBU/LiBr or Triton B gave no or sluggish conversion (data not shown). Typical for stabilized α-phosphonate anions, is that the initial addition to the carbonyl group favors a Z-transition state, while the subsequent rate-determining oxyanion cyclization favors an E-transition state. Reaction parameters such as the selection of the countercation influence energy levels of transition states and reversibility, and therefore, also E/Z selectivity.^19−22^ Changing the reaction solvent to the less coordinating toluene considerably improved the E/Z ratio (entry 5). The same was observed for Et_2_O, although less pronounced (entry 6). DME and MTBE gave lower E/Z ratios (entries 7 and 8), comparable to THF (entry 3). Further enhancing cation solvation by using DMPU as the cosolvent increased the formation of the Z-alkene (entry 9). By lowering the reaction temperature to −78 °C, a very good E/Z ratio of 9:1 was achieved (entry 10). Unfortunately, and unexpectedly, employing a smaller excess of the base and phosphonate 15 gave a diminished stereoselectivity (entry 11). Larger substituents on the phosphonate had no positive effect (entry 12 and 13). While BINOL-based phosphonates occasionally give high Z-selectivity in HWE olefinations,^23^ this preference was not observed in our case (entry 14).
As larger phosphonate substituents did not show a positive influence on the E/Z-selectivity, we aimed at transforming propargyl alcohol 14 into the corresponding dimethylphosphonate 19. Synthesis of the propargylic bromide via an Appel reaction set the stage for phosphonylation by an Arbuzov reaction, effected by refluxing in trimethyl phosphite (Scheme 4). This method furnished phosphonate 19 on multigram-scale in an excellent yield.
To our delight, transferring the optimized HWE olefination conditions to the synthesis of trien-yne alcohol 8 resulted in a satisfying yield and very good E-selectivity (E/Z > 93:7) after one-pot desilylation. Trien-yne alcohol 8 was treated with neat tetramethyldisilazane resulting in the corresponding dimethylsilyl ether, serving as the substrate for a 7-endo-dig hydrosilylation. The course of intramolecular alkyne hydrosilylations depends on the employed catalyst: While Pt and most Ru catalysts effect exo cyclizations,^11,24^ the cationic Ru complex [Cp*Ru(MeCN)3]PF_6_ has been reported to give endo cyclization products with high selectivity.^25,26^ Mechanistically, DFT calculations^27^ indicate an endo transition state similar to 20. Subsequent hydride insertion is accompanied by C–C bond rotation, placing the hydride anti to the silyl group as in simplified transition state 21, thereby establishing the observed trans selectivity. Reductive elimination leads to oxasilacycloheptene 7 with 90% yield from trien-yne alcohol 8. Finally, desilylation of 7 by treatment with TBAF yielded (R)-all-trans-13,14-dihydroretinol (2). The analytical data matched to those previously reported. This synthesis route enables an unprecedented efficient preparation of (R)-all-trans-13,14-dihydroretinol (2), omitting tedious stereocenter formation and carbon skeleton assembly as in the previous published synthesis.^6^
Having established synthetic access to (R)-all-trans-13,14-dihydroretinol (2), we explored its transformation into its main oxidized metabolites. Careful selection of oxidation conditions was required here to keep the sensitive tetraene unit intact. DMP-oxidation of 2 gave (R)-all-trans-13,14-dihydroretinal (4) successfully (Scheme 5). Treatment with in situ formed Ag(I) oxide, known to not affect alkenes,^28^ furnished (R)-all-trans-13,14-dihydroretinoic acid (6), showing gratifying compatibility.
In conclusion, we developed an efficient and stereoselective total synthesis of the vitamin A metabolite (R)-all-trans-13,14-dihydroretinol (2), with an optimized E-selective HWE olefination and a Ru(II) catalyzed intramolecular 7-endo-dig hydrosilylation as the key steps. Furthermore, we have found compatible conditions for stepwise oxidation to (R)-all-trans-13,14-dihydroretinoic acid (6). We are convinced that this novel access to these molecules will facilitate the elucidation of their biological function in future studies.
Experimental Section
General Experimental Information
All reactions were carried out under a nitrogen atmosphere using standard Schlenk techniques. Reaction temperatures refer to the temperature of the heating mantle or cooling bath. Anhydrous solvents (MTBE, CH_2_Cl_2_, THF, toluene) were taken from an MBraun solvent purification system (SPS 800). Other anhydrous solvents were purchased from Sigma-Aldrich or Fisher Scientific and were used without further purification. Aqueous solutions are saturated if not mentioned otherwise. TLC analysis was performed on silica gel 60/Kieselguhr F254, 0.25 mm (Merck). Compounds were visualized using 254 nm UV light followed by KMnO_4_ stain. ^1^H, ^13^C{^1^H} and ^31^P NMR spectra were recorded on an Agilent 400 NMR spectrometer at 400, 101, and 162 MHz, respectively, using CDCl_3_ as the solvent. Chemical shifts are reported in ppm with the solvent resonance as the internal standard (for CDCl_3_: δ 7.26 ppm for ^1^H, δ 77.16 ppm for ^13^C). Data are reported as follows: chemical shifts (δ), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, b = broad, m = multiplet), coupling constant J (Hz), and integration value. High resolution mass spectra (HRMS) were recorded on a Thermo Scientific Orbitrap Exploris 480 mass spectrometer with electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) in positive mode. Specific rotations were determined with a Schmidt
- Haensch Polartronic MH8 polarimeter in a 100 mm path-length cell.
Synthetic Procedures and Analytical Data
(R)-all-trans-13,14-Dihydroretinol (2)
To oxasilacycloheptene 7 (590 mg, 1.7 mmol, 1.0 equiv) in anhydrous THF (20 mL) at 0 °C was added TBAF (1 M in THF, 2.6 mL, 2.6 mmol, 1.5 equiv) and the reaction mixture was stirred for 2 h at r.t. Aqueous NH_4_Cl (20 mL) was added and the aqueous layer was extracted with Et_2_O (3 × 20 mL). The combined organic layers were washed with brine (30 mL) and dried over Na_2_SO_4_. Purification by flash column chromatography (pentane/Et_2_O = 8:2) gave (R)-all-trans-13,14-dihydroretinol 2 as a faint yellow oil (435 mg, 1.5 mmol, 88%).
TLC: Rf = 0.45 (pentane/Et_2_O = 6:4). ^1^H NMR (CDCl_3_, 400 MHz): δ = 6.41 (dd, J = 14.9, 11.1 Hz, 1 H), 6.11 (d, J = 16.2 Hz, 1 H), 6.04 (d, J = 16.2 Hz, 1 H), 5.99 (d, J = 11.1 Hz, 1 H), 5.60 (dd, J = 15.0, 8.4 Hz, 1 H), 3.70–3.61 (m, 2 H), 2.41 (sept, J = 7.1 Hz, 1 H), 2.00 (t, J = 6.8 Hz, 2 H), 1.90 (s, 3 H), 1.69 (s, 3 H), 1.65–1.55 (m, 4 H), 1.49–1.41 (m, 2 H), 1.06 (d, J = 6.7 Hz, 3 H), 1.01 (s, 6 H) ppm. ^13^C{^1^H} NMR (CDCl_3_, 101 MHz): 140.1, 138.0, 137.9, 134.4, 129.7, 129.0, 126.3, 125.8, 61.5, 40.0, 39.7, 34.5, 34.4, 33.1, 29.1, 21.8, 21.1, 19.4, 12.7 ppm. HRMS (ESI) m/z for [M + H]^+^ calculated for C_20_H_33_O 289.2526; found 289.2525. [α]D^20^ = −52 (c 0.1, CHCl_3_).
(R)-all-trans-13,14-Dihydroretinal (4)
To (R)-all-trans-13,14-DHR 2 (40 mg, 0.14 mmol, 1.0 equiv) and pyridine (11 μL, 0.14 mmol, 1.0 equiv) in anhydrous CH_2_Cl_2_ (2.5 mL) at 0 °C was added DMP (88 mg, 0.21 mmol, 1.5 equiv) and the reaction mixture was stirred for 1 h at r.t.. Direct purification by flash column chromatography (pentane/Et_2_O = 98:2) gave (R)-all-trans-13,14-dihydroretinal 4 as a faint yellow oil (32 mg, 0.11 mmol, 81%).
TLC: Rf = 0.50 (pentane/Et_2_O = 95:5). ^1^H NMR (CDCl_3_, 400 MHz): δ = 9.72 (b, 1 H), 6.41 (dd, J = 14.9, 11.2 Hz, 1 H), 6.11 (d, J = 16.1 Hz, 1 H), 6.02 (d, J = 16.1 Hz, 1 H), 5.96 (d, J = 11.0 Hz, 1 H), 5.63 (dd, J = 15.0, 7.6 Hz, 1 H), 2.86 (sept, J = 6.9 Hz, 1 H), 2.47 (ddd, J = 16.3, 7.0, 1.9 Hz, 1 H), 2.39 (ddd, J = 16.3, 6.9, 2.1 Hz, 1 H), 1.98 (t, J = 6.0 Hz, 2 H), 1.88 (s, 3 H), 1.67 (s, 3 H), 1.63–1.53 (m, 4 H), 1.47–1.40 (m, 2 H), 1.10 (d, J = 6.8 Hz, 3 H), 0.99 (s, 6 H) ppm. ^13^C{^1^H} NMR (CDCl_3_, 101 MHz): 202.4, 138.0, 137.8, 137.7, 135.2, 129.3, 129.1, 126.7, 126.2, 50.6, 39.7, 34.4, 33.1, 32.2, 29.1, 21.8, 20.7, 19.4, 12.7 ppm. HRMS (ESI) m/z for [M
- H]^+^ calculated for C_20_H_31_O 287.2369; found 287.2372. [α]D^20^ = −58 (c 0.1, CHCl_3_).
(R)-all-trans-13,14-Dihydroretinoic Acid (6)
AgNO_3_ (41 mg, 0.24 mmol, 2.3 equiv) in water (2 mL) was added dropwise to NaOH (21 mg, 0.52 mmol, 5 equiv) in water (2 mL) at 0 °C. After stirring for 30 min in the dark, (R)-all-trans-13,14-dihydroretinal (4, 30 mg, 0.10 mmol, 1.0 equiv) in THF (0.5 mL) was added dropwise, the cooling bath was removed and stirring was continued for 12 h in the dark. The reaction mixture was subsequently poured into 0.5 M aqueous HCl (10 mL) and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over Na_2_SO_4_. Purification by flash column chromatography (CH_2_Cl_2_/MeOH = 95:5) gave (R)-all-trans-13,14-dihydroretinoic acid 6 as a faint yellow oil (26 mg, 86 μmol, 82%).
TLC: Rf = 0.48 (CH_2_Cl_2_/MeOH = 9:1). ^1^H NMR (CDCl_3_, 400 MHz): δ = 6.43 (dd, J = 14.9, 11.2 Hz, 1 H), 6.11 (d, J = 15.7 Hz, 1 H), 6.02 (d, J = 16.1 Hz, 1 H), 5.96 (d, J = 11.1 Hz, 1 H), 5.63 (dd, J = 15.0, 7.6 Hz, 1 H), 2.84–2.70 (m, 1 H), 2.45–2.25 (m, 2 H), 1.98 (t, J = 6.0 Hz, 2 H), 1.88 (s, 3 H), 1.67 (s, 3 H), 1.63–1.53 (m, 2 H), 1.47–1.40 (m, 2 H), 1.10 (d, J = 6.7 Hz, 3 H), 0.99 (s, 6 H) ppm. ^13^C{^1^H} NMR (CDCl_3_, 101 MHz): 178.1, 138.0, 137.8, 137.7, 135.0, 129.5, 129.1, 126.5, 126.1, 41.5, 39.7, 34.4, 34.0, 33.1, 29.1, 21.8, 20.4, 19.4, 12.7 ppm. HRMS (ESI) m/z for [M
- H]^+^ calculated for C_20_H_31_O_2_ 303.2319; found 303.2318. [α]D^20^ = −46 (c 0.1, CHCl_3_).
(R)-2,2,5-Trimethyl-3-((1E,3E)-2-methyl-4-(2,6,6-trimethylcyclohex-1-en-1-yl)buta-1,3-dien-1-yl)-2,5,6,7-tetrahydro-1,2-oxasilepine
(7)
Trien-yne alcohol 8 (667 mg, 2.3 mmol, 1.0 equiv) was dissolved in 1,1,3,3-tetramethyldisilazane (2.1 mL, 11.6 mmol, 5.0 equiv) and stirred for 18 h at r.t. Most of the solvent was evaporated in vacuo (approximately 4 mbar, 50 °C sand bath temperature) to give the crude dimethylsilyl ether that was taken to the next step without further purification.
The crude dimethylsilyl ether was dissolved in anhydrous CH_2_Cl_2_ (10 mL), cooled to 0 °C and Cp*Ru(MeCN)4_PF_6 (59 mg, 116 μmol, 5 mol %) was added. After stirring for 4 h at r.t., the reaction mixture was diluted with pentane (10 mL) and filtered over a short plug of florisil with the aid of pentane/Et_2_O (98:2). Drying the filtrate in vacuo yielded sufficiently pure oxasilacycloheptene 7 (720 mg, 2.1 mmol, 90%) as a yellow oil.
TLC: Rf = 0.40 (pentane/Et_2_O = 98:2). ^1^H NMR (CDCl_3_, 400 MHz): δ = 6.16–5.83 (m, 4 H), 3.98–3.78 (m, 2 H), 2.80–2.60 (m, 1 H), 1.98 (t, J = 6.0 Hz, 2 H), 1.95–1.88 (m, 1 H), 1.82 (s, 3 H), 1.69 (s, 3 H), 1.64–1.51 (m, 3 H), 1.48–1.40 (m, 2 H), 1.13 (d, J = 7.1 Hz, 3 H), 1.00 (s, 3 H) ppm. ^13^C{^1^H} NMR (CDCl_3_, 101 MHz): 150.8, 139.5, 138.3, 138.0, 134.5, 132.3, 128.7, 125.5, 62.3, 39.7, 38.2, 34.4, 33.3, 33.1, 29.1, 29.1, 22.2, 21.8, 19.4, 13.7, −0.6, −0.9 ppm. HRMS (APCI) m/z for [M + H]^+^ calculated for C_22_H_37_OSi 345.2608; found 345.2611.
(R,6E,8E)-3,7-Dimethyl-9-(2,6,6-trimethylcyclohex-1-en-1-yl)nona-6,8-dien-4-yn-1-ol
(8)
NaHMDS (2 M in THF, 3.1 mL, 6.2 mmol, 2.0 equiv) was added to phosphonate 19 (2.86 g, 6.2 mmol, 2.0 equiv) in anhydrous toluene (24 mL) at −78 °C and the mixture was stirred for 30 min. β-ionone (635 μL, 3.1 mmol, 1.0 equiv) in precooled anhydrous toluene (6 mL) was added dropwise and the reaction mixture was stirred for 1 h. Afterward, TBAF (1 M in THF, 9.4 mL, 9.4 mmol, 3.0 equiv) was added and after warming to r.t., stirring was continued for 5 h. Aqueous NH_4_Cl (20 mL) was added and the aqueous layer was extracted with Et_2_O (3 × 30 mL). The combined organic layers were washed with brine (50 mL) and dried over Na_2_SO_4_. Purification by flash column chromatography (pentane/Et_2_O = 7:3) gave the desired trien-yne alcohol 8 as a yellow oil (667 mg, 2.3 mmol, 75%).
TLC: Rf = 0.34 (pentane/Et_2_O = 8:2). ^1^H NMR (CDCl_3_, 400 MHz): δ = 6.17 (d, J = 15.8 Hz, 1 H), 6.04 (d, J = 16.1 Hz, 1 H), 5.38 (s, 1 H), 3.87–3.75 (m, 2 H), 2.87–2.75 (m, 1 H), 2.03–1.95 (m, 2 H), 2.00 (s, 3 H), 1.79–1.68 (m, 3 H), 1.66 (s, 3 H), 1.62–1.54 (m, 2 H), 1.47–1.40 (m, 2 H), 1.24 (d, J = 6.9 Hz, 3 H), 0.98 (s, 3 H) ppm. ^13^C{^1^H}-NMR (CDCl_3_, 101 MHz): 146.5, 137.6, 135.7, 129.8, 129.0, 108.8, 100.7, 80.2, 61.4, 39.8, 39.6, 34.3, 33.1, 29.0, 24.1, 21.8, 21.6, 19.4, 15.1 ppm. HRMS (APCI) m/z for [M + H]^+^ calculated for C_20_H_31_O 287.2369; found 287.2370. [α]D^20^ = −38 (c 0.1, CHCl_3_).
(R)-4-((tert-Butyldiphenylsilyl)oxy)-2-methylbutan-1-ol
(12)
LiAlH_4_ (2.56 g, 68.1 mmol, 3.0 equiv) was added portionwise to R-(+)-methylsuccinic acid (11, 3.00 g, 22.7 mmol, 1.0 equiv) in anhydrous THF (80 mL) at 0 °C. The mixture was stirred for 1 h at r.t. and then for 15 h at 60 °C by heating with a sand bath. After cooling to r.t., Rochelle salt (38.5 g, 136 mmol, 6 equiv) and water (6 mL) were added and stirring was continued overnight. The mixture was filtered over a short plug of Celite with the aid of THF. The filtrate was dried in vacuo and the crude diol was taken to the next step without further purification.
The crude diol was dissolved in anhydrous DMF (90 mL), cooled to −40 °C and DBU (5.1 mL, 34.1 mmol, 1.5 equiv) was added. Then, TBDPSCl (5.9 mL, 22.7 mmol, 1.0 equiv) was added over 2 h by syringe pump. Stirring was continued for 8 h before quenching the reaction by addition of aqueous NaHCO_3_ (100 mL). The aqueous layer was extracted with Et_2_O (3 × 100 mL) and the combined organic layers were washed with brine (200 mL). Drying over Na_2_SO_4_ and purification by flash column chromatography (pentane/Et_2_O = 8:2) afforded the desired alcohol 12 (5.14 g, 15.0 mmol, 66% over two steps) as a colorless oil. The analytical data matched to those previously reported.^29^
(R)-4-((tert-Butyldiphenylsilyl)oxy)-2-methylbutanal
(13)
Et_3_N (8.3 mL, 60.0 mmol, 4.0 equiv) and pyridine-SO_3_ (4.78 g, 30.0 mmol, 2.0 equiv) were added to alcohol 12 (5.14 g, 15.0 mmol, 1.0 equiv) in anhydrous CH_2_Cl_2_ (120 mL) and anhydrous DMSO (30 mL) at 0 °C. After stirring for 1 h at r.t., aqueous NH_4_Cl (120 mL) was added and the aqueous layer was extracted with CH_2_Cl_2_ (3 × 100 mL). The combined organic layers were washed with brine (200 mL) and dried over Na_2_SO_4_. Purification by flash column chromatography (pentane/Et_2_O = 98:2) yielded the desired aldehyde 13 as a colorless oil (4.79 g, 14.1 mmol, 94%). The analytical data matched to those previously reported.^30^
(R)-6-((tert-Butyldiphenylsilyl)oxy)-4-methylhex-2-yn-1-ol
(14)
CBr_4_ (7.79 g, 23.5 mmol, 2.0 equiv) was added portionwise to aldehyde 13 (4.00 g, 11.7 mmol, 1.0 equiv) and PPh_3_ (12.3 g, 47.0 mmol, 4.0 equiv) in CH_2_Cl_2_ (70 mL) at 0 °C. After stirring for 1 h at r.t., most of the solvent was evaporated and the residue diluted with pentane (150 mL). Filtration over a short plug of SiO_2_ with the aid of pentane and drying of the filtrate in vacuo gave the desired dibromoalkene that was taken to the next step without further purification.
To the crude dibromoalkene in anhydrous THF (50 mL) at −78 °C was added nBuLi (1.6 M in hexane, 16.2 mL, 25.8 mmol, 2.2 equiv) dropwise. The reaction mixture was stirred for 1.5 h and then warmed to 0 °C. After stirring for 30 min, paraformaldehyde (0.81 g, 27.0 mmol, 2.3 equiv) was added, the ice-bath was removed and stirring was continued for 15 h. Aqueous NH_4_Cl (50 mL) was added and the aqueous layer was extracted with Et_2_O (3 × 50 mL). The combined organic layers were dried over Na_2_SO_4_. Purification by flash column chromatography (pentane/Et_2_O = 8:2) yielded the desired propargyl alcohol 14 as a colorless oil (3.52 g, 9.6 mmol, 82% over two steps).
TLC: Rf = 0.37 (pentane/Et_2_O = 8:2). ^1^H NMR (CDCl_3_, 400 MHz): δ = 7.74–7.64 (m, 4 H), 7.47–7.35 (m, 6 H), 4.19 (d, J = 2.0 Hz, 2 H), 3.86–3.72 (m, 2 H), 2.82–2.72 (m, 1 H), 1.77–1.60 (m, 2 H), 1.43 (s, 1 H), 1.17 (d, J = 7.0 Hz, 3 H), 1.06 (s, 9 H) ppm. ^13^C{^1^H} NMR (CDCl_3_, 101 MHz): δ = 135.8*, 135.7*, 134.02*, 133.97*, 129.7, 127.73*, 127.71*, 90.5, 78.8, 61.7, 51.5, 39.6, 27.0, 22.5, 21.0, 19.3 ppm. HRMS (APCI) m/z for [M + H]^+^ calculated for C_23_H_31_O_2_Si 367.2088; found 367.2088. [α]D^20^ = −40 (c 0.1, CHCl_3_).
*1:1 splitted ^13^C NMR signals are partially observed for aromatic carbons in the TBDPS substituent. This indicates hindered rotation of the silyl ether, leading to rotamers.
Dibenzyl (3-(trimethylsilyl)prop-2-yn-1-yl)phosphonate (Bn-15)
NaHMDS (2 M in THF, 1.0 mL, 2.0 mmol, 1.0 equiv) was added to dibenzyl phosphite (0.44 mL, 2.0 mmol, 1.0 equiv) in anhydrous THF (3 mL) at −10 °C and the mixture was stirred for 15 min. (3-bromo-prop-1-ynyl)-trimethylsilane (0.33 mL, 2.0 mmol, 1.0 equiv) was added and stirring was continued for 1 h. Then, the reaction was quenched by addition of water (10 mL) and the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic layers were washed with aqueous HCl (2 M, 30 mL) and water (30 mL). After drying over Na_2_SO_4_, purification by flash column chromatography (pentane/EtOAc = 7:3) gave the desired phosphonate Bn-15 as a colorless oil (490 mg, 1.3 mmol, 66%).
TLC: Rf = 0.23 (pentane/EtOAc = 8:2). ^1^H NMR (CDCl_3_, 400 MHz): δ = 7.39–7.28 (m, 10 H), 5.18–5.03 (m, 4 H), 2.84 (s, 1 H), 2.79 (s, 1 H), 0.13 (s, 9 H) ppm. ^13^C{^1^H} NMR (CDCl_3_, 101 MHz): 136.1 (d, J = 6.2 Hz), 128.7, 128.6, 127.9, 95.5 (d, J = 14.3 Hz), 88.4 (d, J = 8.8 Hz), 68.5 (d, J = 6.6 Hz), 19.8 (d, J = 144.8 Hz), – 0.1 (d, J = 1.3 Hz) ppm. ^31^P NMR (CDCl_3_, 162 MHz): 21.8 (s) ppm. HRMS (ESI) m/z for [M + Na]^+^ calculated for C_20_H_25_O_3_PSiNa 395.1203; found 395.1199.
(4S)-4-(3-(trimethylsilyl)prop-2-yn-1-yl)dinaphtho[2,1-d:1′,2′-f][1,3,2]dioxaphosphepine
4-oxide (BINOL-15)
To rac-BINOL (500 mg, 1.7 mmol, 1.0 equiv) in anhydrous CH_2_Cl_2_ (12 mL) at 0 °C was added Et_3_N (0.63 mL, 4.5 mmol, 2.6 equiv) and methyl dichlorophosphite (0.20 mL, 2.1 mmol, 1.2 equiv) successively. The mixture was stirred for 12 h while warming to r.t.. Et_2_O (50 mL) was added and the resulting suspension was filtered over Celite with Et_2_O washings. Evaporation in vacuo yielded the crude phosphite that was taken to the next step without further purification.
The crude phosphite was dissolved in 3-bromo-1-(trimethylsilyl)-1-propyne (2.5 mL) and stirred at 160 °C by heating with a sand bath for 2 h. After cooling to r.t., direct purification by flash column chromatography (pentane/Et_2_O = 1:1) gave the desired phosphonate BINOL-15 as an off-white foam (170 mg, 0.4 mmol, 22% over two steps).
TLC: Rf = 0.17 (pentane/Et_2_O = 6:4). ^1^H NMR (CDCl_3_, 400 MHz): δ = 8.02 (dd, J = 8.9, 4.4 Hz, 2 H), 7.93 (d, J = 8.2 Hz, 2 H), 7.59 (dd, J = 8.9, 0.8 Hz, 1 H), 7.55 (d, J = 8.9 Hz, 1 H), 7.50–7.43 (m, 2 H), 7.35 (d, J = 8.3 Hz, 1 H), 7.33–7.26 (m, 3 H), 3.19–2.97 (m, 2 H), 0.01 (s, 9 H) ppm. ^13^C{^1^H} NMR (CDCl_3_, 101 MHz): 147.8 (d, J = 10.5 Hz), 145.8 (d, J = 10.0 Hz), 132.6 (d, J = 1.3 Hz), 132.5 (d, J = 1.6 Hz), 132.0 (d, J = 1.4 Hz), 131.8 (d, J = 1.2 Hz), 131.5 (d, J = 1.5 Hz), 131.3 (d, J = 0.7 Hz), 128.7 (d, J = 0.6 Hz), 128.5 (d, J = 0.5 Hz), 127.4, 127.1, 127.0, 126.9, 126.0, 126.0 (d, J = 0.5 Hz), 121.9 (d, J = 2.6 Hz), 121.6 (d, J = 2.1 Hz), 121.1 (d, J = 2.1 Hz), 120.4 (d, J = 3.7 Hz), 93.0 (d, J = 14.4 Hz), 90.0 (d, J = 9.2 Hz), 17.8 (d, J = 141.0 Hz), −0.30 (d, J = 1.2 Hz) ppm. ^31^P NMR (CDCl_3_, 162 MHz): 30.0 (s) ppm. HRMS (ESI) m/z for [M + H]^+^ calculated for C_26_H_24_O_3_PSi 443.1227; found 443.1228.
Dimethyl (R)-(6-((tert-butyldiphenylsilyl)oxy)-4-methylhex-2-yn-1-yl)phosphonate
(19)
PPh_3_ (1.79 g, 6.8 mmol, 1.0 equiv) was added to propargyl alcohol 14 (2.50 g, 6.8 mmol, 1.0 equiv) and CBr_4_ (2.26 g, 6.8 mmol, 1.0 equiv) in anhydrous CH_2_Cl_2_ (50 mL) at 0 °C. After stirring for 10 min at the same temperature, additional 3 × 0.1 equiv of PPh_3_ were added every 5 min. The reaction mixture was diluted with pentane (100 mL) and filtered over a small plug of SiO_2_ with the help of pentane/Et_2_O (98:2). Drying the filtrate in vacuo gave the crude propargyl bromide that was taken to the next step without further purification.
The crude propargyl bromide was refluxed in P(OMe)3 (15 mL) by heating with a sand bath for 12 h. Afterward, the excess of P(OMe)3 was removed by rotary evaporation at 70 °C. The residue was purified by flash column chromatography (pentane/EtOAc = 4:6) to yield the desired phosphonate 19 as a colorless oil (2.85 g, 6.2 mmol, 91% over two steps).
TLC: Rf = 0.43 (pentane/EtOAc = 4:6). ^1^H NMR (CDCl_3_, 400 MHz): δ = 7.69–7.61 (m, 4 H), 7.44–7.32 (m, 6 H), 3.82–3.69 (m, 8 H), 2.76–2.63 (m, 1 H), 2.72 (d, J = 2.0 Hz, 1 H), 2.67 (d, J = 2.2 Hz, 1 H), 1.69–1.60 (m, 2 H), 1.13 (d, J = 6.8 Hz, 3 H), 1.02 (s, 9 H) ppm. ^13^C{^1^H} NMR (CDCl_3_, 101 MHz): 135.70*, 135.66*, 134.1*, 134.0*, 129.70*, 129.69*, 127.74*, 127.73*, 87.5 (d, J = 10.2 Hz), 69.4 (d, J = 14.6 Hz), 53.6 (d, J = 6.8 Hz), 39.7 (d, J = 2.8 Hz), 26.9, 22.6 (d, J = 2.9 Hz), 21.0 (d, J = 3.1 Hz), 19.3, 17.2 (d, J = 146.5 Hz) ppm. ^31^P NMR (CDCl_3_, 162 MHz): 25.0 (s) ppm. HRMS (ESI) m/z for [M + Na]^+^ calculated for C_25_H_35_O_4_PSiNa 481.1934; found 481.1933. [α]D^20^ = −12 (c 0.05, CHCl_3_).
*1:1 splitted ^13^C NMR signals are observed for aromatic carbons in the TBDPS substituent. This indicates hindered rotation of the silyl ether, leading to rotamers.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Beltrán-de-Miguel B.; Estévez-Santiago R.; Olmedilla-Alonso B. Assessment of dietary vitamin A intake (retinol, α-carotene, β-carotene, β-cryptoxanthin) and its sources in the National Survey of Dietary Intake in Spain (2009–2010). Int. J. Food Sc. Nutr. 2015, 66 (6), 706–712. 10.3109/09637486.2015.1077787.26313699 · doi ↗ · pubmed ↗
- 2Menezes M. S. S.; Almeida C. M. M. Structural, functional, nutritional and clinical aspects of vitamin A: A review. Pharma Nutrition 2024, 27, 10038310.1016/j.phanu.2024.100383. · doi ↗
- 3Álvarez R.; Vaz B.; Gronemeyer H.; de LeraÁ. R. Functions, Therapeutic Applications, and Synthesis of Retinoids and Carotenoids. Chem. Rev. 2014, 114, 1–125. 10.1021/cr 400126 u.24266866 · doi ↗ · pubmed ↗
- 4Moise A. R.; Kuksa V.; Imanishi Y.; Palczewski K. Identification of All-trans-Retinol:All-trans-13,14-dihydroretinol Saturase. J. Biol. Chem. 2004, 279 (48), 50230–50242. 10.1074/jbc.M 409130200.15358783 PMC 2665716 · doi ↗ · pubmed ↗
- 5Moise A. R.; Isken A.; Domínguez M.; de Lera A. R.; von Lintig J.; Palczewski K. Specificity of Zebrafish Retinol Saturase: Formation of All-trans-13,14-dihydroretinol and All-trans-7,8- dehydroretinol. Biochemistry 2007, 46, 1811–1820. 10.1021/bi 062147 u.17253779 PMC 2561287 · doi ↗ · pubmed ↗
- 6Moise A. R.; Dominguez M.; Alvarez S.; Alvarez R.; Schupp M.; Cristancho A. G.; Kiser P. D.; de Lera A. R.; Lazar M. A.; Palczewski K. Stereospecificity of Retinol Saturase: Absolute Configuration, Synthesis, and Biological Evaluation of Dihydroretinoids. J. Am. Chem. Soc. 2008, 130, 1154–1155. 10.1021/ja 710487 q.18179220 PMC 2621334 · doi ↗ · pubmed ↗
- 7Weber P.; Flores R. E.; Kiefer M. F.; Schupp M. Retinol Saturase: More than the Name Suggests. Trends Pharmacol. Sci. 2020, 41 (6), 418–427. 10.1016/j.tips.2020.03.007.32345479 · doi ↗ · pubmed ↗
- 8Moise A. R.; Kuksa V.; Blaner W. S.; Baehr W.; Palczewski K. Metabolism and transactivation activity of 13,14-dihydroretinoic acid. J. Biol. Chem. 2005, 280 (30), 27815–27825. 10.1074/jbc.M 503520200.15911617 PMC 1352314 · doi ↗ · pubmed ↗