Genome and antibiotic resistance characteristics of Shigella clinical isolates in Fujian Province, Southeast China, 2005–2019
Mengying Huang, Xiaoxuan Zhang, Chaochen Luo, Haibin Xu, Yufeng Qiu, Jinsong Yang

TL;DR
This study analyzed the genome and antibiotic resistance of Shigella isolates in Fujian, China, finding high multidrug resistance and identifying key resistance genes and genetic structures.
Contribution
The study provides a detailed genomic characterization of multidrug-resistant Shigella isolates in Fujian, revealing resistance gene patterns and phylogenetic clusters.
Findings
All Shigella isolates were multidrug-resistant, with high resistance to cefotaxime, ciprofloxacin, and azithromycin.
blaCTX-M-14 and blaCTX-M-15 genes were common, with specific genetic environments and plasmid types identified.
Phylogenomic analysis showed Fujian isolates were mostly endemic, not linked to major international outbreak lineages.
Abstract
Shigellosis is a serious public health issue in many developing countries. The emergence of multidrug-resistant (MDR) Shigella isolates has deepened the treatment difficulty and health burden of shigellosis. China is the largest developing country in the world, but so far, the genome of MDR Shigella isolates has not been well characterized. In this study, 60 clinical isolates of Shigella spp. in Fujian Province, southeast China, from 2005 to 2019 were characterized for drug resistance phenotype, whole-genome sequencing and bioinformatics analysis. The results showed that the MDR rate of Shigella isolates was 100%, among which the resistance rates of cefotaxime, ciprofloxacin and azithromycin were 36.67, 21.67 and 10.00 %, respectively. The positive rate of extended-spectrum beta-lactamase (ESBL)-producing strains was 23.33%. The resistance profiles of Shigella flexneri and Shigella…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
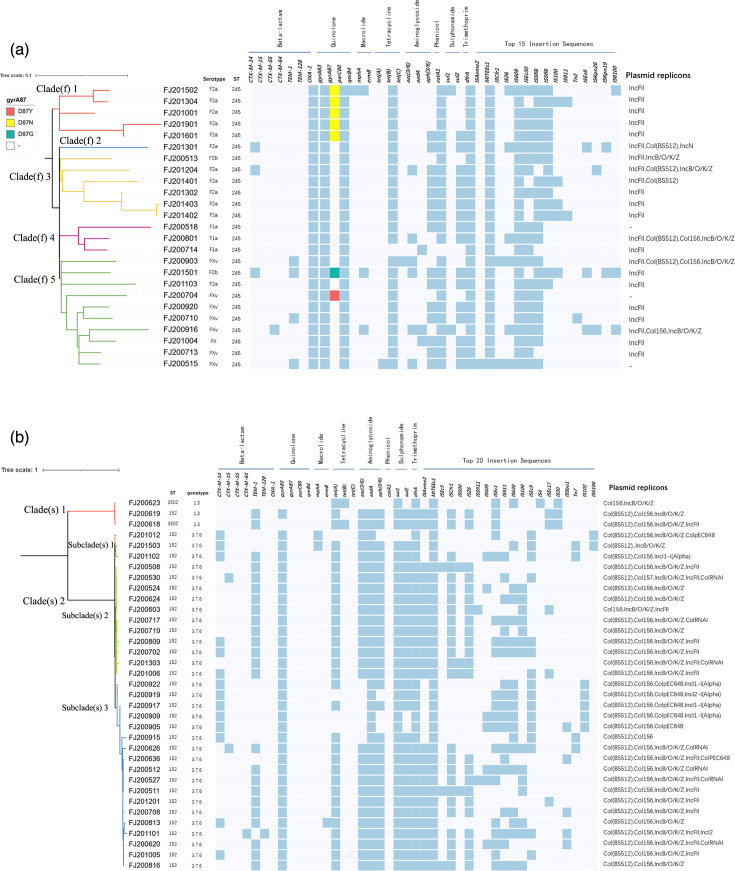 Fig. 1
Fig. 1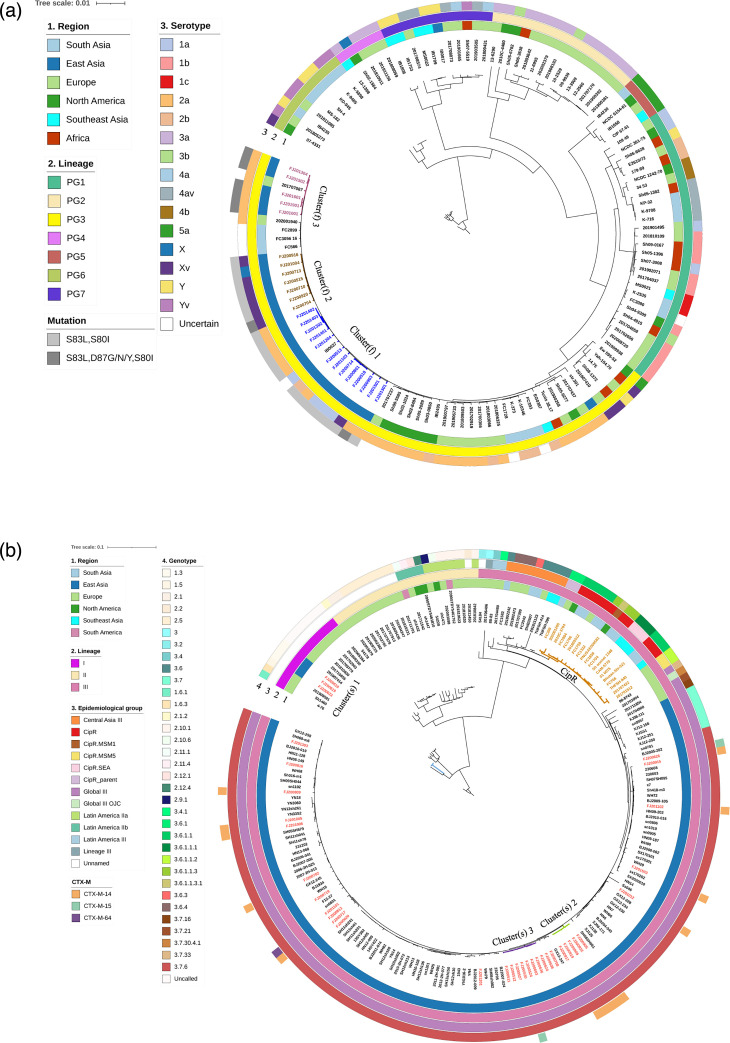 Fig. 2
Fig. 2| Antibiotic | Total ( |
| |||
| AMP | 100 | 100.00 | 100.00 | – | – |
| CTX | 36.67 | 28.00 | 42.86 | 1.386 | 0.239 |
| CAZ | 3.33 | 4.00 | 2.86 | 0.059 | 1.000 |
| CZO | 43.33 | 32.00 | 51.43 | 2.242 | 0.134 |
| CFX | 1.67 | 4.00 | 0.00 | 1.424 | 0.417 |
| FEP | 10 | 12.00 | 8.57 | 0.011 | 0.917 |
| IPM | 3.33 | 4.00 | 2.86 | 0.059 | 1.00 |
| NAL | 100.00 | 100.00 | 100.00 | – | – |
| CIP | 23.33 | 52.00 | 2.86 | 19.688 | <0.001 |
| LEV | 13.33 | 8.00 | 5.71 | 0 | 1.000 |
| GEN | 55.00 | 16.00 | 77.14 | 21.832 | <0.001 |
| CHL | 35.00 | 84.00 | 0.00 | 45.231 | <0.001 |
| STR | 100.00 | 100.00 | 100.00 | – | – |
| TET | 83.33 | 92.00 | 77.14 | 1.371 | 0.242 |
| SMZ | 76.67 | 68.00 | 82.86 | 1.799 | 0.180 |
| SIZ | 80.00 | 68.00 | 88.57 | 3.857 | 0.049 |
| AZM | 8.33 | 8.00 | 8.57 | 0 | 1.000 |
| PMB | 0.00 | 0.00 | 0.00 | – | – |
| ESBLs | 23.33 | 16 | 28.57 | 1.288 | 0.256 |
| Antibiotics* | Phenotype: non-susceptible†(no. of isolates) | Phenotype: susceptible(no. of isolates) | Sensitivity (%) | Specificity (%) | Major resistance genes | ||
| Genetype resistant | Genetype susceptible | Genetype resistant | Genetype susceptible | ||||
| AMP | 59 | 1 | 0 | 0 | 98.33 |
| |
| CTX | 16 | 6 | 5 | 33 | 72.73 | 86.84 |
|
| CAZ | 2 | 0 | 20 | 38 | 100 | 65.51 |
|
| NAL | 58 | 2 | 0 | 0 | 96.67 | ||
| CIP | 25 | 2 | 0 | 33 | 92.53 | 100.00 | |
| AZM | 6 | 0 | 0 | 54 | 100.00 | 100.00 | |
| SIZ | 44 | 4 | 6 | 6 | 91.67 | 50.99 | |
| GEN | 29 | 4 | 4 | 23 | 87.88 | 85.19 | |
| TET | 51 | 3 | 4 | 2 | 94.44 | 33.33 | |
| CHL | 23 | 0 | 0 | 37 | 100.00 | 100.00 | |
| Overall | 313 | 22 | 39 | 226 | 93.43 | 85.28 | |
- —http://dx.doi.org/10.13039/501100017686 Fujian Provincial Health Technology Project
- —http://dx.doi.org/10.13039/501100003392 Natural Science Foundation of Fujian Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEscherichia coli research studies · Viral gastroenteritis research and epidemiology · Salmonella and Campylobacter epidemiology
Data Summary
The short-read sequence data have been submitted to the National Center for Biotechnology Information National Library of Medicine under BioProject accession number PRJNA1111321. The detailed genomic information of Fujian isolates and the global isolates used in this study can be found in Data S1 and S2, available in the online version of this article, respectively.
Introduction
Shigellosis is one of the most common diarrhoeal diseases in the world, leading to an estimated 1.88 trillion infections and 1.1 million deaths annually. Seventy per cent of these deaths are related to children under 5 years old and immunocompromised patients [12]. Shigella spp. is the causative agent of shigellosis in humans. There are four species within the Shigella genus, which include Shigella dysenteriae, Shigella boydii, Shigella flexneri and Shigella sonnei, with the latter two being the predominant serogroups responsible for more than 90% of all bacillary dysentery cases worldwide [3]. The distribution and changes in Shigella composition vary by country, region and year. It was reported that S. sonnei affects the adult population of economically developed countries, while S. flexneri remains the predominant pathogenic agent in childhood diarrhoea in low- to middle-income countries. Nevertheless, with development, there is a progressive decrease in the detection of S. flexneri and an increase in S. sonnei [46]. Shigellosis is also an important public health problem in China; although its incidence is decreasing year by year, it is still higher than in many developed countries [78]. Surveillance data showed that the main isolated strains in China were S. flexneri and S. sonnei. Except for Beijing, S. flexneri was most prevalent in central and northern China, while S. sonnei was most prevalent in the east and south [7]. Fujian Province, located on the southeast coast of China with convenient transportation, is a core area of the ‘21st Century Maritime Silk Road’. With economic and social development, exchanges between Fujian and other countries are becoming closer. Although the incidence of shigellosis in Fujian is relatively low, the potential risk of Shigella transmission is increasing.
In the past, treatment for shigellosis relied on older antimicrobial drugs, such as tetracycline, chloramphenicol, ampicillin, trimethoprim–sulfamethoxazole and nalidixic acid [9]. However, due to selection pressure and the widespread use of mobile elements associated with resistance, multidrug-resistant (MDR) strains to these antibiotics are a common problem. In recent years, the World Health Organization recommended ciprofloxacin as the first choice for the treatment of shigellosis in adults and children, with ceftriaxone, pivmecillinam and azithromycin as the second choice [10]. Unfortunately, strains of Shigella resistant to antibiotics, especially ciprofloxacin, third-generation cephalosporins and azithromycin, have been found in Europe, America, Asia and many other countries [1114]. International travel and sexual transmission among men who have sex with men (MSM) contribute to the spread of Shigella between countries and become a potential risk for the emergence of MDR strains [15].
Compared to traditional methods like antibiotic susceptibility testing and PCR detection of drug resistance genes, whole-genome sequencing offers several advantages for obtaining antimicrobial resistance (AMR) gene data. Whole-genome sequencing can simultaneously detect all genes and mutations, discover possible new resistance determinants, reveal resistance mechanisms and distinguish strains with the same resistance pattern caused by different mechanisms. Additionally, it provides reliable basic information on microorganisms, clear phylogenetic relationships and comprehensive information for epidemiological investigations [1620]. In addition, genome-wide data can be stored indefinitely and analysed repeatedly at any time. Open databases such as ResFinder and the Comprehensive Antibiotic Resistance Database (CARD) also provide simple and easy-to-learn methods for predicting resistance genes [2123].
So far, limited research exists on comparing whole-genome characteristics and drug resistance phenotypes of Shigella in specific regions of China, particularly regarding population structure and transmission routes. To address this gap, we aimed to contribute to the data on drug resistance and molecular epidemiology of Shigella in Fujian Province. We collected 60 Shigella strains isolated from 2005 to 2019 and performed drug resistance and phylogenetic analysis based on whole genome sequencing. The findings of this study provide a basis for effectively preventing and controlling shigellosis, conducting scientific monitoring, guiding clinical medication rationally and controlling the spread of MDR strains.
Methods
Study population and Shigella isolates
From 2005 to 2019, a total of 60 Shigella isolates were obtained from clinical stool samples of 60 patients (duplicate isolates from the same individual were excluded) with acute bacillary dysentery admitted to diarrhoeic clinics in four surveillance districts in Fujian Province, Southeast China. The patients’ medical records were reviewed to collect their demographic characteristics. Cases of adults with chronic diarrhoea were not included in this study. All the isolates were confirmed according to a standard protocol [24], and serological confirmation was performed by slide agglutination test with polyvalent and monovalent antisera to Shigella (Denka Seiken, Chuo-ku, Japan) following the manufacturer’s instructions.
Antimicrobial susceptibility testing
The broth dilution method was used to detect the minimum inhibitory concentration (MIC) of 18 antibiotics using the Sensititre AIM system (Thermo Fisher, USA) and customized antimicrobial susceptibility test (AST) plate Gov1 and Gov2 drug susceptibility culture plates (Thermo Fisher) following the manufacturer’s instructions (http://www.trekds.com/techinfo/). The antibiotics tested and MIC ranges included ampicillin (AMP, 2–64), tetracycline (TET, 1–32), chloramphenicol (CHL, 2–64), sulfamethoxazole (SMZ, 0.25/4.75 to 8/152), cefazolin (CZO, 0.5–16), cefotaxime (CTX, 0.25–8), ceftazidime (CAZ, 0.25–8), cefoxitin (CFX, 2–64), gentamicin (GEN, 1–32), imipenem (IMI, 0.25–8), nalidixic acid (NAL, 2–64), azithromycin (AZI, 2–64), sulfisoxazole (SIZ, 32–512), ciprofloxacin (CIP, 0.03–32), levofloxacin (LEV, 0.12–8), polymyxin B (PMB, 0.5–16), cefepime (FEP, 0.25–16) and streptomycin (STR, 4–32). Escherichia coli ATCC 25922 was used as a quality control. The Clinical and Laboratory Standards Institute (CLSI) guideline M100-S31 was used for the determination of AMR break points and for the screening and confirmation of extended-spectrum beta-lactamases (ESBLs) [25]. Owing to the absence of a clinical break point for azithromycin in CLSI, the epidemiological cut-off values of azithromycin for Shigella was considered to be 16 mg l^−1^ according to the European Committee on Antimicrobial Susceptibility Testing break points [26]. MDR was defined as the emergence of resistance to three or more classes of antibiotics.
Whole-genome sequencing and processing
Bacterial genomic DNA was extracted from pure broth cultures of Shigella isolates using a Bacterial Genomic DNA extraction kit (51 304, QIAGEN, Germany) according to the manufacturer’s instructions. After quality control, a 350-bp DNA fragment library was constructed using the Nextera XT v2 kit (Illumina, San Diego, USA) and sequenced for 150-bp paired-end reads on an Illumina HiSeq 2500 platform (Illumina). On average, 500 Mb of clean data was generated per strain, with a minimum sequencing depth of 150×. FastQC (v0.11.5, https://www.bioinformatics.babraham.ac.uk/projects/fastqc), SPAdes (v3.14) [27] and Prokka (v1.14.6) [28] software were used for quality control, de novo assembly and annotation of the sequencing data, respectively. Additionally, we representatively selected six bla_CTX-M-14_, bla_CTX-M-15_ and mphA-harbouring isolates for long-read genome sequencing using an Oxford Nanopore MinION sequencer. Canu v1.6 [29] was used for de novo assemblies, and Pilon v1.24 [30] was applied to correct the sequencing errors by the Illumina sequencing data.
Identification of resistance genes, insertion sequences and plasmids
Two free analysis websites, ResFinder (https://cge.food.dtu.dk/services/ResFinder) and CARD (https://card.mcmaster.ca/analyze/rgi), were used to predict the presence and mutations of antibiotic resistance genes and mobile genetic elements (MGEs) including plasmids. ResFinder utilizes a blast search engine with a 90% identity threshold and 60% coverage rate. The presence of insertion sequences (ISs) and plasmids was analysed using MobileElementFinder v1.0.3 (https://cge.food.dtu.dk/services/MobileElementFinder/) and PlasmidFinder v2.0.1 (https://cge.food.dtu.dk/services/PlasmidFinder/), respectively, with a discrimination threshold of 95% for both tools.
Genotyping and population structure analysis
ShigaPass (version 1.5.0) was applied for serotype confirmation of S. flexneri (https://github.com/imanyass/ShigaPass) [31]. Genotyping of S. sonnei was performed by Sonneityping tool (version 20210201) with the hierarchical single-nucleotide variant-based genotyping scheme (https://github.com/katholt/sonneityping) and implemented in Mykrobe software version 0.9.0 [4]. The E. coli/Shigella EnteroBase-specific scheme cgMLST protocol was used to analyse the Shigella population structure. The cgMLST alleles obtained by comparison with 2513 single-copy orthologous genes were used to calculate the individual genetic distance between the genomes (https://enterobase.warwick.ac.uk), and the population structure was visualized using minimum-spanning tree [32].
Phylogenetic analysis
Roary-based phylogenetic analysis
After genome annotation by Prokka v1.14.6 [28], core and accessory genes were identified and extracted. Roary v3.13.0 [33] was then used to compare the identified core genes. A maximum likelihood phylogenetic tree was generated by FastTree v2.1.10 with the General Time-Reversible (GTR) nucleotide substitution model to assess relatedness among isolate genomes, followed by visualization and annotation with ITOL v6.0 (https://itol.embl.de/).
SNP-based phylogenetic analysis
To understand the global phylogenetic placement of Fujian Shigella isolates, maximum likelihood phylogenetic trees were drawn based on 131 genomes of S. flexneri (local 25 isolates and 106 globally distributed isolates) and 204 genomes of * S. sonnei* (local 35 isolates and 169 globally distributed isolates), respectively, as detailed in Data S2. Briefly, a pseudo genome was constructed for each sample by substitution with the alternative allele at variant sites in the BIM Collaboration Format (BCF) file in the reference genome (S. flexneri 2a strain 301: GenBank entry number CP000038 and S. sonnei Ss046: NC_004337.2). Then, a multi-FASTA whole genome alignment of 25 S. flexneri and 35 S. sonnei samples was generated, respectively. The contig files were added to the alignment with Snippy v4.4.5 (https://github.com/tseemann/snippy), using the ‘–ctgs’ flag. Putative recombination regions were detected and masked with Gubbins v2.3.4 [34], and a maximum-likelihood phylogenetic tree was constructed using FastTree (v2.1.10) under the GTR substitution model. The tree layout was graphically edited and annotated using ITOL v6.0 (https://itol.embl.de/).
Statistical analysis
SPSS statistical software (version 23) was used for data analysis. The chi-square test was employed to determine significant differences between variables. A P value of less than 0.05 was considered statistically significant.
Results
Demographic data of the patients
Among the patients (n=60, median age 33.6 years, ranged 1–83), the male–female ratio was 1.61 : 1 (37 vs. 23), 22 patients (36.67%) were younger than 5, 15 patients (25.00%) were aged 5–14, 10 patients (16.67%) were aged 15–65 and 13 (21.67 %) were older than 65. All the patients showed acute diarrhoea symptoms, followed by abdominal pain (85.0%), vomiting (68.3%), fever (61.67%), rectal tenesmus (46.7%) and nausea (28.3%). All the patients recovered after treatment.
Serotype and genotype of the isolates
The 60 Shigella isolates collected during 2005–2019 were identified as 25 S. flexneri and 35 S. sonnei by slide agglutination test, and the identity of serotype by ShigaPass was consistent with that of slide agglutination test. In the 25 strains of * S. flexneri*, serotype 2a was the predominant serotype among the isolates (48.0%), followed by serotype X variant (Xv) (28.0%), 1a (12.0%), 2b (8%) and X (4.0%). As for genotyping of S. sonnei, most belonged to genotype 3.7.6 of Lineage III, except for two isolates classified as genotype 1.3 of Lineage I.
Phenotype analysis of antibiotic resistance
All Shigella isolates exhibited MDR. The distribution of resistance classes showed that 8.33, 6.67, 55.00 and 30.00% of isolates were resistant to 3, 4, 5, and 6 classes of antimicrobials, respectively. Notably, all isolates were resistant to ampicillin, nalidixic acid and streptomycin but remained sensitive to polymyxin B. There were significant differences in resistance rates between the two Shigella species, as detailed in Table 1. S. flexneri had higher resistance rates to ciprofloxacin (P<0.001) and chloramphenicol (P<0.001) than S. sonnei, and the opposite was observed for gentamicin resistance (P<0.001). The rates of S. flexneri and S. sonnei being resistant to one or more first-line antibiotics (third-generation cephalosporin, ciprofloxacin and azithromycin) were 100% (25/25) and 51.43% (18/35), respectively. About 23.33% (14/60) of the Shigella isolates produced ESBLs, of which S. flexneri included Xv (n=3) and F1a (n=1), and S. sonnei were all genotypes 3.7.6 (n=10). The resistance rates of S. sonnei to cefotaxime and azithromycin increased significantly from 2005~ to 2015~ (from 33.33 to 100% for cefotaxime and from 3.7 to 100% for azithromycin), while the resistance rates of S. flexneri to ciprofloxacin and azithromycin increased more obvious from 2005~ to 2015~ (from 27.27 to 75.00% for ciprofloxacin and from 9.09 to 50.00% for azithromycin; Fig. S1).
Determinants of resistance
Genomic analysis of the 60 Shigella isolates revealed a diverse array of AMR determinants. A total of 18 different resistance genes or mutations were identified across seven classes of antibiotics, including beta-lactams (bla_TEM_, bla_OXA_, bla_CTX-M_), quinolones (gyrA, parC, qnrB4), macrolides (mphA, ermB), tetracyclines (tetA, tetB, tetC), aminoglycosides (aac(3/6), aadA, aph(3/6)), chloramphenicol (catA1) and sulfamethoxazole (sul1, sul2, dfrA; Table S1). Additionally, 30 ISs and 16 different plasmid replicon types were found in the isolates.
β-Lactam resistance genes
The major beta-lactam resistance genes identified in the 60 Shigella isolates were the penicillin resistance genes (bla_OXA_, bla_TEM_) and the ESBL-encoding gene (bla_CTX-M_). Importantly, bla_OXA_ was mainly found in S. flexneri (n=25), all of which were bla_OXA-1_, and bla_TEM_ was mainly found in S. sonnei (n=35), all of which were bla_TEM-1_ except one bla_TEM-128_. S. sonnei harboured bla_CTX-M-14_ (n=14), bla_CTX-M-15_ (n=2) and bla_CTX-M-64_ (n=1), and S. flexneri carried bla_CTX-M_ included bla_CTX-M-14_ (n=3) and bla_CTX-M-55_ (n=1; Data S1). The proportion of S. sonnei carrying bla_CTX-M_ gene (17/35) was higher than that in S. flexneri (4/25), P<0.05.
Quinolone resistance genes
Quinolone resistance-determining region (QRDR) mutations, including gyrA and parC, were found in a high proportion (e.g., >80 %) of the Shigella isolates. Four non-synonymous point mutations were detected in the gyrA gene*,* including codon 83 (Ser to Leu, S83L) and codon 87 (Asp to Asn, D87N*, Asp to Tyr,* D87Y and Asp to Gly, D87G). Only the Ser to Ile (S80I) point mutation was detected in parC gene (Data S1). Four mutation patterns, including gyrA (S83L) + parC (S80I), gyrA (D87N, S83L) + parC (S80I), gyrA (D87G, S83L) + parC (S80I) and gyrA (D87Y, S83L) + parC (S80I), were identified in S. flexneri, with the first being the predominant mutation pattern (n=18). In contrast, S. sonnei isolates harboured only the gyrA (S83L) mutation. The plasmid-mediated quinolone resistance (PMQR) gene qnrB was also found in S. flexneri, while none of the PMQR genes were detected in S. sonnei. The prevalence of these mutations is detailed in Fig. 1.
Midpoint-rooted phylogenetic tree of 25 S. flexneri isolates (a) and 35 S. sonnei isolates (b) based on the Roary analysis, and the distribution of AMR determinants. Clades(f) and Clades(s)/Subclade(s) labelled in different colours indicated the clades of S. flexneri and S. sonnei, respectively. Blue boxes represented the identity of different types of AMR genes and ISs. GyrA mutations, including D87Y, D87N and D87G, were separately indicated by the colours red, yellow and green. Serotype, multi-locus sequence typing (MLST) and plasmid replicons of the isolates were also listed.
Macrolide resistance genes
Macrolide resistance genes identified in Shigella included mphA and ermB. The mphA gene displayed a broader presence, detected in both S. flexneri (n=3) and S. sonnei (n=2) isolates. In contrast, the ermB gene was found exclusively in a single S. sonnei strain (Fig. 1).
Tetracycline, aminoglycosides, chloramphenicol and sulfonamides resistance genes
There were significant differences in the distribution of tetracycline resistance genes tet(A, B) between S. flexneri and S. sonnei (P< 0.001). S. flexneri displayed a higher prevalence of tetB (n=24) compared to tetA (n=1) and tetC (n=1), whereas only tetA (n=30) and tetB (n=2) were detected in S. sonnei (Fig. 1). aac(3/6), aadA(3/6) and aph(3/6) were predominant aminoglycoside resistance genes in Shigella, whose proportion in S. sonnei (28/35, 30/35, 34/35) was higher than that in S. flexneri (6/25, 15/25, 3/25), with P values <0.001, <0.05 and <0.001, respectively. The chloramphenicol resistance gene catA1 was mainly found in S. flexneri (n=24). No S. sonnei harboured this gene. The main sulfamethoxazole resistance genes detected in S. flexneri were sul2 (n=16) and dfrA, including dfrA1 (n=25), dfrA12 (n=2), dfrA14 (n=2) and dfrA17 (n=2), and the main sulfamethoxazole resistance genes detected in S. sonnei were sul1 (n=34), sul2 (n=31) and dfrA, including dfrA1 (n=33), dfrA12 (n=24) and dfrA17 (n=1) (Fig. 1).
MGEs associated with AMR
Notably, all 60 Shigella strains harboured the IS MITEEc1. The top five IS elements in S. flexneri were MITEEc1 (n=25), IS609 (n=25), ISSf18 (n=22), ISEc30 (n=20) and ISSf19 (n=14). Only one isolate carried the Tn2 transposon. In contrast, S. sonnei displayed a wider variety of IS elements. The top eight identified were MITEEc1 (n=35), ISAeme2 (n=25), ISCfr1 (n=25), IS26 (n=22), ISEc1 (n=20), IS630 (n=19), ISEc9 (n=16) and IS911 (n=11). Four strains harboured Tn7 transposons. Except for three isolates lacking a plasmid replicon, S. flexneri harboured four types of plasmid replicon, with IncF (n=22) being the most dominant. In contrast to S. flexneri, S. sonnei displayed a higher diversity with eight identified replicon types. Notably, Col (BS512) (n=33), Col156 (n=33), IncB/O/K/Z (n=28) and IncF (n=15) were the main types found in S. sonnei (Fig. 1).
Genetic environment of blaCTX−M
Genetic environment surrounding the bla_CTX−M_ genes can be divided into five types and named type I–V (Data S1), including type I (n=11) ‘ISEcp1 -bla_CTX−M_ -IS903’, type II (n=7) ‘ISEcp1 -bla_CTX−M_’, type III (n=1) ‘ISEcp1 -bla_CTX−M_ -ORF477’, type IV (n=1) ‘ISEcp1 -bla_CTX−M_ -ΔIS903’ and type V (n=1) ‘IS26 -ISEcp1 -bla_CTX−M_ -IS903’. The mobile element ISEcp1 was consistently found upstream of bla_CTX−M_ genes, and IS903 (complete or incomplete) was mainly included downstream of the genes. Among them, type I and type II were the predominant (38.10%, 8/21 and 33.33 %, 7/21) genetic environment of the bla_CTX−M−14_ gene and plasmids containing these structures included IncFII, IncI1, IncI2 and IncN.
Consistency analysis of drug resistance phenotype and genotype
In this study, 10 commonly used antibiotics were selected to analyse the correlation between antibiotic resistance phenotypes and genotypes. A total of 600 phenotypic results were obtained from 60 Shigella isolates, of which 539 results exhibited phenotype–genotype consistency (overall consistency rate: 89.83%). Among the 331 resistant phenotypes, 22 lacked detectable drug-resistant genes or mutations, resulting in an overall sensitivity of 93.43%. Six isolates displayed discrepancies for β-lactams, primarily cefotaxime. The specificity of the genotypic analysis, which refers to the proportion of truly susceptible isolates lacking resistance genes, was 85.28% (Table 2). Of the 265 isolates with phenotypic sensitivity, 39 harboured drug-resistant genes or mutations. Twenty-one isolates with β-lactam resistance genes showed sensitivity to ceftazidime.
Population structure analysis
The genetic structure of the S. flexneri and S. sonnei bacterial population was determined by the EnteroBase cgMLST scheme, and a minimum spanning tree was constructed and demonstrated, respectively (Fig. S2), which were compared with the classification results of Roary analysis. All S. flexneri isolates belonged to a single sequence type, ST245 (n=25), but the genetic distances among the five serotypes of S. flexneri presented distant genetic relationships (with an average of 86 different alleles). The S. flexneri strains classified as the same clade in Fig. 1a can be further distinguished by cgMLST analysis and exhibited distinct genetic distances, such as Clade(f) 1 (FJ201001, FJ201304, FJ201502, FJ201601 and FJ201901 of F2a) and Clade(f) 4 (FJ200518, FJ200801 and FJ200714 of F1a). The * S. sonnei* isolates were divided into two clusters according to the genotype 3.7.6 (n=32) and 1.3 (n=3), consistent with Figs 1b and S2. The S. flexneri and S. sonnei isolates with the same genotypic drug-resistant profile could be classified as the same cluster, within 20 allele differences.
SNP-based phylogenetic analysis
The phylogenetic tree of S. flexneri (Fig. 2a) showed that the 131 genomes were divided into seven distinct phylogenetic groups (PGs), and the Fujian isolates were all distributed in PG 3. The S. flexneri isolates in Fujian could be subdivided into three clusters, named as Cluster(f) 1, 2 and 3, and Cluster(f) 1 was the largest, containing nine isolates of 2a, three of 1a and one of Xv, exhibiting 5–191 SNPs. These isolates appeared in the 10-year period between 2005 and 2015, showing the long-term colonization characteristics of S. flexneri species. In addition, serotypes 2a, 1a and Xv were dispersed in Cluster(f) 1, indicating several independent serotype conversions. There were 7, 1 and 4 isolates of ciprofloxacin-resistant in Cluster(f) 1, 2 and 3, respectively, and F2a accounted for 66.67% (8/12). Nearly all isolates in the three clusters carried gyrA S83L and parC S80I mutations, but Cluster(f) 3 carried more D87N/G/Y mutations. Fujian isolates of Cluster 1 showed a close relationship to a domestic strain of China (IB0037), while Cluster 3 was clustered with an isolate from France (201707087), suggesting that these two clusters may have different evolutionary origins. Moreover, the results of phylogenetic classification based on SNP and Roary analysis were similar, for example, Cluster(f) 3 was consistent with Clade(f) 1 (including 5 F2a), while Cluster(f) 2 matched several strains in Clade(f) 5 (including 6 FXv and 1 Fx) (Figs1a2a).
Phylogenetic tree for core-genome SNP of S. flexneri isolates (a) and S. sonnei isolates (b) in a global context. Genome 07–4331 and 4–76 were used as out-group to root the trees of S. flexneri and S. sonnei, respectively. Cluster(f) and Cluster(s) indicated the clades of S. flexneri and S. sonnei, respectively. Blue, orange and peach branches denoted Cluster(f) 1–3, while light blue, green and purple represented Cluster(s) 1–3, and gold for ciprofloxacin resistance (CipR) epidemiology groups. The annotations (a) were (from inner to outer ring): region of the isolates, PGs, serotype and QRDR mutation in gyrA and parC. The annotations (b) were (from inner to outer ring): region of the isolates, Lineage, epidemiological group, genotype and blaCTX-M gene type.
For S. sonnei, the 35 genomes can be ascribed to two lineages, among which Lineage I contained three strains with genotype 1.3 (named Cluster(s) 1), and Lineage III contained 32 strains with genotype 3.7.6, including two major groups (named Cluster(s) 2 and 3) (Fig. 2b), and the classification was identical to the population structure analysis (Figs 1b and S2B). Cluster(s) 2 and Cluster(s) 3 were the largest genetic branch with 7–141 SNPs, and six strains carried the bla_CTX-M_ genes (5 of bla_CTX-M-14_ and 1 of bla_CTX-M-15_). According to the classification of the epidemiological group, Cluster(s) 2 and Cluster(s) 3 belonged to Global III, which was genetically distant from the global ciprofloxacin resistance (CipR) groups, such as CipR_parent, CipR MSM5 and CipR_SEA. Except for Cluster(s) 1, the Fujian isolates and the majority of the Chinese isolates with genotype 3.7.6 were all located in Lineage III and formed a distinct Chinese subgroup.
Discussion
The widespread use of antimicrobials has led to the emergence of MDR Shigella, posing a serious threat to the effective treatment of bacillary dysentery and becoming a significant burden on global public health. Currently, ciprofloxacin, azithromycin and ceftriaxone are the most commonly used antibiotics for the empirical treatment of shigellosis [1]. Worryingly, resistance to azithromycin, ciprofloxacin and third-generation cephalosporins is rising globally among Shigella strains [43536]. This study revealed that all Shigella isolates from Fujian Province were MDR, with a higher incidence compared to the reported rate in Asia (68.7%, 95 % CI: 59.9–77.5) [36]. The strains displayed 100% resistance to ampicillin, nalidixic acid and streptomycin, indicating the inadequacy of these traditional antibiotics for treating shigellosis. In our region, the resistance rates of S. flexneri and S. sonnei isolates to cefotaxime from 2005 to 2019 reached 28% (7/25) and 42.86% (15/35), respectively, which were similar to the resistance rates of S. flexneri (34.40%) and * S. sonne*i (43.2%) reported in Jiangsu Province of China from 2012 to 2015 [37]. The positive rate of ESBL-producing Shigella isolates in Fujian was 23.33% (14/60), similar to the positive rate in Jiangsu Province of China from 2013 to 2015 (25.89 %) [37]. Although this rate was higher than that reported in Africa (1.5%) [38], it was still lower than the rate observed in southwest Iran (43.0%) [39] and Barcelona of Spain (28.75%) [40]. The non-susceptible rate to ciprofloxacin of Shigella isolates in Fujian was 43.33% (26/60), which was comparable to the national resistance rate of 44.65% reported in China in 2014 [7]. It is noteworthy that the resistance rate of S. flexneri to ciprofloxacin in Fujian was 52.00%, significantly higher than that of S. sonnei and similar to the rate observed in Taiyuan, Shanxi Province [41]. In this study, S. flexeri Xv had high resistance to first-line antibiotics, among which the positive rate of ESBL-producing strains was 42.86% (3/7), the non-sensitivity rate to ciprofloxacin was 100% and the resistance rate to azithromycin was 14.29% (1/7). S. flexneri Xv is a newly identified serotype that is currently widespread in China, and it has also acquired resistance to a variety of antibiotics [42]. The emergence of new serotypes with enhanced resistance to first-line antibiotics may be a strategy for S. flexneri to maintain its epidemic capacity. The resistance rates of Shigella isolates in Fujian to the three first-line antibiotics showed upward trends in general, especially the rates of S. flexneri to ciprofloxacin and S. sonnei to cefotaxime, and it was similar to the global pattern [4344]. Antibiotic usage in China is complex and widespread, and China has become the largest producer and consumer of antibiotics in the world [45]. Among them, cephalosporins, macrolides, fluoroquinolones and penicillins are the most commonly used antibiotics in China, not only for clinical treatment but also for the livestock industry [4647]. These antibiotics may cause persistent selection pressure in the clinic, agriculture and environment, leading to the emergence and spread of severe MDR Shigella in China.
Historically, public health efforts to monitor and address bacterial AMR relied primarily on low-resolution phenotypic analyses. However, the affordability of whole genome sequencing is revolutionizing this approach by providing a high-resolution view of the evolution and spread of AMR [48]. Building upon drug resistance phenotypes, further understanding of the epidemiological significance of resistance-determining factors and their ability to predict drug resistance is crucial. In the present study, 18 different drug-resistant genes across seven classes of antibiotics were found in the Shigella isolates of Fujian. Among the β-lactam resistance genes identified, bla_TEM_, bla_OXA_ and bla_CTX-M_ were present. The genetic environment of most bla_CTX-M-14_ included an intact ISEcp1 42 bp upstream of the bla_CTX-M-14_ open reading frame and IS903 downstream, while the genetic environment of bla_CTX-M-15_ consisted of ISEcp1 -bla_CTX−M_ -ORF477, and the results were consistent with those of S. sonnei isolates in the Republic of Korea [49]. Fluoroquinolones, such as ciprofloxacin, are first-line antibiotics for the treatment of shigellosis. The high ciprofloxacin non-susceptible rate (MIC ≥0.5 mg l^−1^) observed in S. flexneri (100%, 25/25) compared to S. sonnei (5.71%, 2/35) can be attributed to differences in the QRDR. In S. flexneri, the QRDR displayed a combination pattern with dual mutations of the gyrA gene (one pattern was S83L, the other was D87G, D87N or D87Y) and a single mutation of the parC gene (S80I) [50]. In contrast, only a single mutation of the gyrA (S83L) gene was detected in S. sonnei. In China, in addition to the most common S83L mutation of the gyrA gene, D87N and D87G mutations have been reported in the eastern provinces [5152]. Interestingly, the D87Y mutation was detected only in S. flexneri isolated from calf faeces in a northwestern province, which suggests that Shigella isolates of animal origin are a potential risk for the spread of fluoroquinolone resistance [53].
The consistency analysis between genotype and phenotype of Shigella antibiotic resistance revealed an overall coincidence rate of 88.50%. The genotypic antimicrobial susceptibility testing demonstrated sensitivity and specificity of 93.35 and 82.53 %, respectively. However, a few discrepancies between the phenotype and genotype were observed for certain antibiotics. For instance, isolates harbouring the bla_CTX-M_ gene remained susceptible to cefotaxime and ceftazidime. The genome analysis of these isolates has reported disruption of the promoter regions of bla_CTX-M_ due to the inversion of an IS26 element [3540]. Additionally, variations in promoter activity due to different spacer sequences can also influence bla_CTX-M_ expression [54].
Phylogenetic analysis allows for the comparison of local Shigella genome data with global population data, providing insights into the life cycle and evolutionary relationships of Shigella isolates. Connor’s [55] study classified S. flexneri into seven PGs, with PG1, PG2, PG4 and PG6 considered the oldest lineages, while PG3 and PG5 are more recent. In this study, all S. flexneri isolates from Fujian Province (2005–2019) belonged to PG3 lineages, forming locally colonized populations with genetic diversity. The ability of independent serotype conversions, such as serotype 2a, 1a and Xv dispersed in cluster(f) 1, might contribute to the long-term success of S. flexneri locally [55]. Fujian cluster(f) 3 was clustered with an isolate from France (201707087). These findings indicated that the epidemic of S. flexneri in Fujian was caused by both local expansion and interregional transmission. For S. sonnei, five distinct lineages have been identified globally, with Lineage III emerging as the dominant lineage worldwide over the past two decades [4]. Clades 3.6 and 3.7 are the most common (19 and 62 %, respectively) and account for the majority of S. sonnei in all continents except Latin America [4]. In our study, the majority of S. sonnei isolates in Fujian Province belonged to Lineage III with genotype 3.7.6 and clustered with the majority of the Chinese isolates, forming a distinct Chinese clade (main Chinese clade) [56]. The phylogenetic tree revealed that Fujian isolates were genetically distant from the recent emergence of MDR clones of central Asian sub-lineage III (with genotypes 3.6.0, 3.6.2, 3.6.3 and 3.6.4) and CipR (with genotype 3.6.1.1 and its sub-genotypes), which are considered as part of a global pandemic [12]. These differences in the genetic distance might explain the low resistance to ciprofloxacin of Fujian S. sonnei at gene level. The Fujian isolates carrying multiple types of drug resistance genes (such as bla_CTX-M_ gene) were widely distributed in the China clade, suggesting that the MDR S. sonnei existed in co-circulation domestically. Notably, bla_CTX-M-14_ and bla_CTX-M-15_ were the dominant bla_CTX-M_ types identified in this study, contrasting with the increasing prevalence of bla_CTX-M-27_ observed in developed countries [5759]. The heterogeneous geographical distribution of bla_CTX-M_ may be attributed to the differences in circulating clones between regions.
This study has several limitations that should be acknowledged. First, the selection of 60 representative Shigella isolates from Fujian Province may have limited the captured diversity of genome features. Moreover, due to self-medication practices in mild bacillary dysentery cases, it was not possible to obtain and analyse the genome characteristics of Shigella isolates associated with diarrhoea in this population. Second, due to the lack of detailed epidemiological background data and the difficulty of conducting retrospective investigations, substantial information about cases (such as travel information) might be subject to omissions and mistakes, so it could not provide epidemiological evidence for the determination of international imports.
In conclusion, this study provided insights into the antibiotic resistance of Shigella and the AMR determinants they carried in Fujian Province, Southeast China. The results showed that the prevalence of MDR Shigella was significant in Fujian Province, and the resistance rates to first-line antibiotics were on the rise. There was high consistency between AMR determinants and antibiotic resistance phenotypes. The antibiotic resistance of Shigella to cefotaxime, azithromycin and ciprofloxacin may be driven by a blaCTX-M- and mphA(ermB)-encoding plasmid and double/triple mutations in the QRDR, respectively. It also revealed a high degree of genetic diversity in the circulating Shigella strains, and there may be possible interaction between S. flexneri and international strains, while S. sonnei was dominated by local circulating clones, belong to China-specific clade. Continuous surveillance and further genomic epidemiological studies with diverse sources of Shigella strains are urgently needed to provide the basis for transmission risk assessment, clinical treatment and vaccine strategies.
supplementary material
10.1099/mgen.0.001325Uncited Table S1.
10.1099/mgen.0.001325Uncited Fig. S1.
10.1099/mgen.0.001325Uncited Supplementary Data Sheet 1.
10.1099/mgen.0.001325Uncited Supplementary Data Sheet 2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kotloff KL Riddle MS Platts-Mills JA Pavlinac P Zaidi AKM Shigellosis Lancet 201839180181210.1016/S 0140-6736(17)33296-829254859 · doi ↗ · pubmed ↗
- 2Rolfo F Marin GH Silberman M Pattin J Giugnio S et al Epidemiological study of shigellosis in an urban area of Argentina J Infect Dev Ctries 2012632432810.3855/jidc.197722505441 · doi ↗ · pubmed ↗
- 3Gu B Cao Y Pan S Zhuang L Yu R et al Comparison of the prevalence and changing resistance to nalidixic acid and ciprofloxacin of Shigella between Europe-America and Asia-Africa from 1998 to 2009 Int J Antimicrob Agents 20124091710.1016/j.ijantimicag.2012.02.00522483324 · doi ↗ · pubmed ↗
- 4Hawkey J Paranagama K Baker KS Bengtsson RJ Weill F-X et al Global population structure and genotyping framework for genomic surveillance of the major dysentery pathogen, Shigella sonnei Nat Commun 202112268410.1038/s 41467-021-22700-433976138 PMC 8113504 · doi ↗ · pubmed ↗
- 5Thompson CN Duy PT Baker S The rising dominance of Shigella sonnei: an intercontinental shift in the etiology of bacillary dysentery P Lo S Negl Trop Dis 20159 e 000370810.1371/journal.pntd.000370826068698 PMC 4466244 · doi ↗ · pubmed ↗
- 6Torraca V Holt K Mostowy S Shigella sonnei Trends Microbiol 20202869669710.1016/j.tim.2020.02.01132663462 PMC 7611039 · doi ↗ · pubmed ↗
- 7Chang Z Zhang J Ran L Sun J Liu F et al The changing epidemiology of bacillary dysentery and characteristics of antimicrobial resistance of Shigella isolated in China from 2004-2014 BMC Infect Dis 20161668510.1186/s 12879-016-1977-127863468 PMC 5116132 · doi ↗ · pubmed ↗
- 8Kotloff KL Winickoff JP Ivanoff B Clemens JD Swerdlow DL et al Global burden of Shigella infections: implications for vaccine development and implementation of control strategies Bull World Health Organ 19997765166610516787 PMC 2557719 · pubmed ↗