Genome-wide analyses of Mycobacterium tuberculosis complex isolates reveal insights into circulating lineages and drug resistance mutations in The Gambia
Leopold Tientcheu, Fatou Faal, Naffie Top, Olimatou Jobe, Sang Marie Colley, Abigail Ayorinde, Alieu Mendy, Binta Sarr-Kuyateh, Simon Donkor, Martin Antonio, Bouke de Jong, Andrea Rachow, Beate Kampmann, Jayne S. Sutherland, Hongwei Li, Tom Blundell, Susana Campino, Thomas Kohl

TL;DR
This study uses genome sequencing to analyze TB strains in The Gambia, revealing lineage diversity and drug resistance patterns to improve local TB control.
Contribution
The study provides new insights into MTBC lineage distribution and drug resistance mutations specific to The Gambia.
Findings
Lineage 4 and 6 strains dominate in The Gambia, with lineage 4 showing higher genetic variability.
A significant proportion of isolates had uncertain drug resistance mutations, including a lineage 6-specific ethambutol mutation.
Drug resistance mutations often occur in conserved, solvent-inaccessible protein regions, affecting stability and fitness.
Abstract
Tuberculosis (TB), caused by the Mycobacterium tuberculosis complex (MTBC), remains a pressing global health challenge, with the West African region, including The Gambia, experiencing a substantial burden. This study explores the genetic diversity of MTBC strains circulating in The Gambia for nearly two decades (2002–2021) to enhance understanding of drug resistance dynamics and inform targeted diagnostic and treatment strategies. Using whole-genome sequencing (WGS) data from 1,803 TB isolates, we identified the predominance of lineage 4 (L4, 67.2%) and lineage 6 (L6, 26.6%) strains, with L4 showing more significant genetic variability over time. Drug susceptibility analysis of these isolates revealed that 78% (1421 isolates) were drug-susceptible, while 6.5% (119 isolates) exhibited resistance, primarily to isoniazid, rifampicin, and their combination. Additionally, 15.5% (282…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
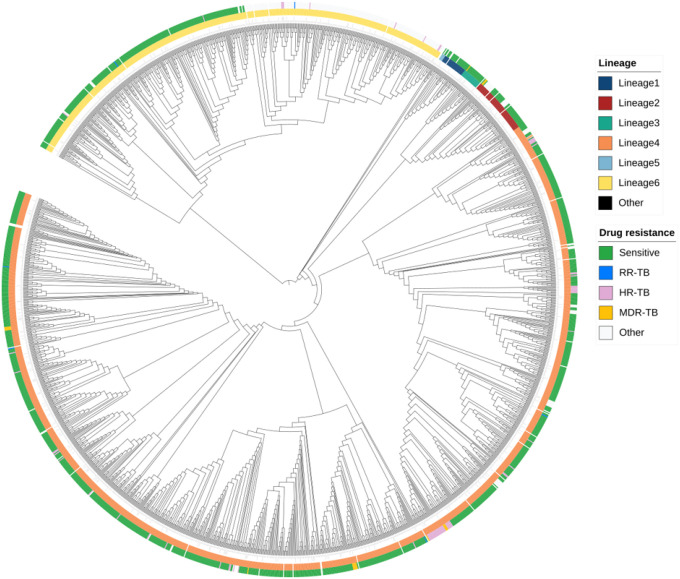 Figure 1
Figure 1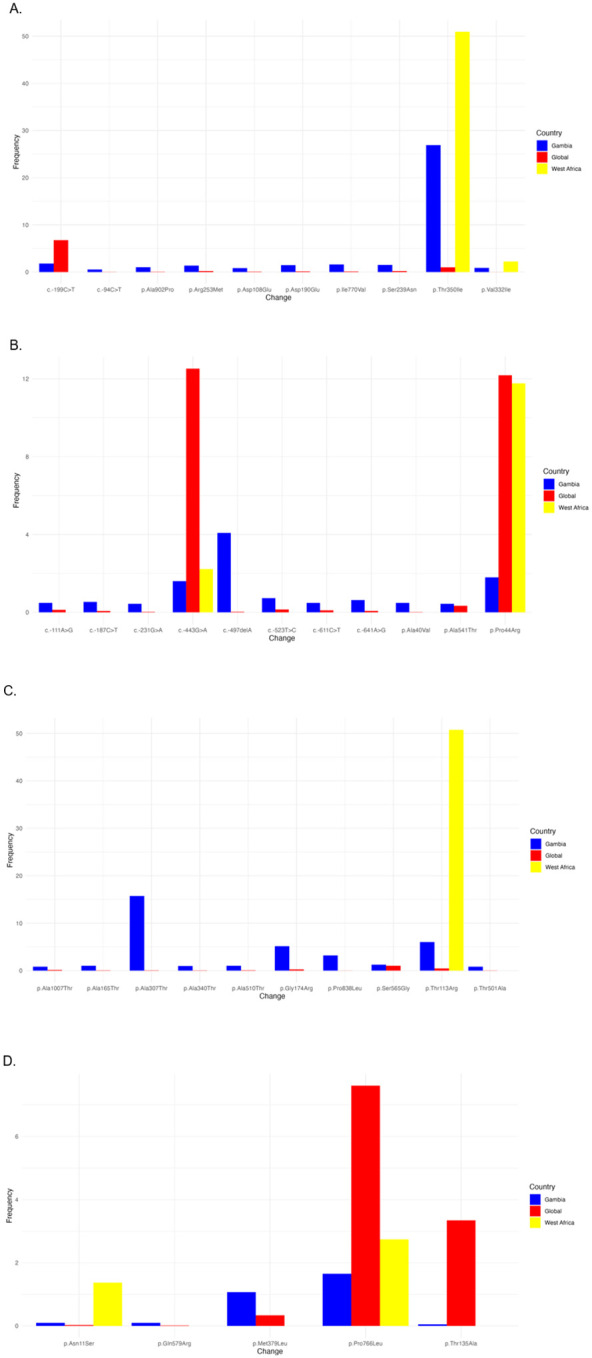 Figure 2
Figure 2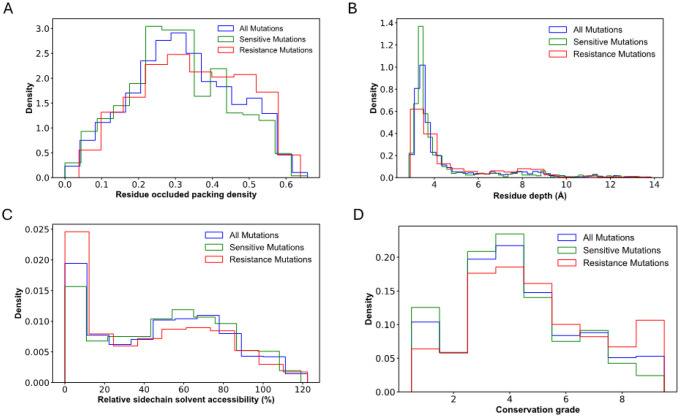 Figure 3
Figure 3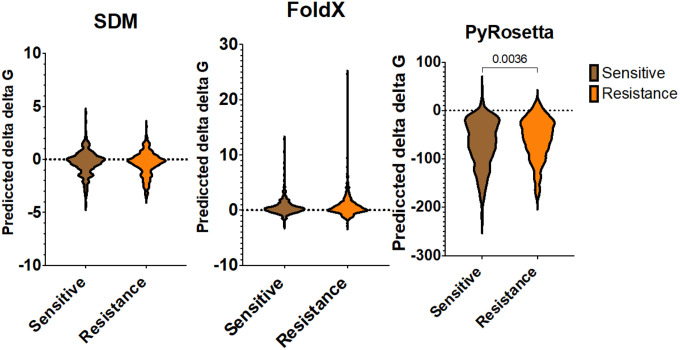 Figure 4
Figure 4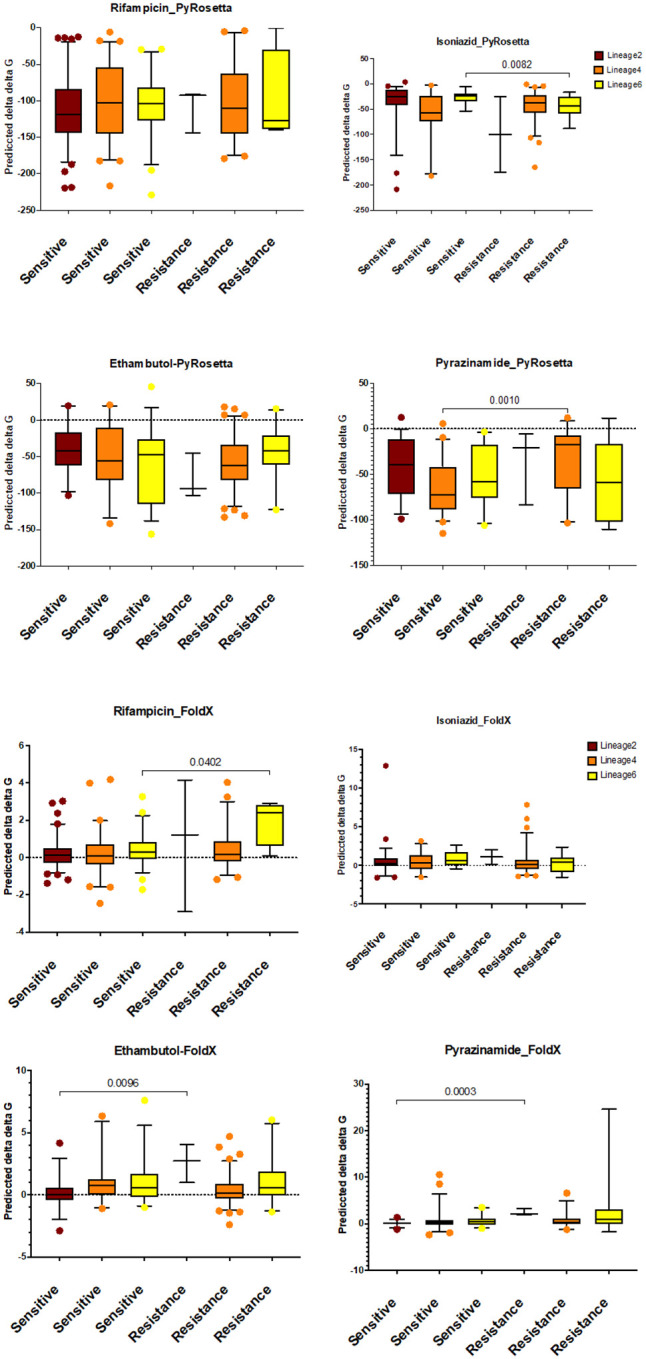 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTuberculosis Research and Epidemiology · Mycobacterium research and diagnosis · vaccines and immunoinformatics approaches
Introduction
Tuberculosis (TB) caused by bacilli of the Mycobacterium tuberculosis complex (MTBC) remains a significant public health problem worldwide. In 2022, an estimated 10.6 million people developed TB, with 1.3 million deaths reported. West Africa accounted for 10% of global TB deaths in 2022, including 3,900 cases and 580 fatalities in The Gambia. TB disproportionately affects resource-limited communities and low-income countries ^1^.
Whole genome sequencing (WGS) advances have greatly enhanced our understanding of MTBC lineage diversity and phylogeographical distribution ^2, 3^. Of the ten known MTBC lineages (L1-L10) (Guyeux et al., 2024), all are found in Africa. In West Africa, TB is driven by M. tuberculosis sensu stricto (Mtb) and M. africanum (Maf) lineages, which co-exist in affected populations ^4^. While geographically restricted lineages like M. africanum (L5, L6) are endemic to West Africa, globally disseminated lineages such as Mb-Beijing (L2) and Mb-Europe-America (L4) are also prevalent (Galagan et al., 2014).
West Africa follows the World Health Organization (WHO) guidelines for TB treatment, involving a prolonged multi-drug regimen ^1^. TB patients’ treatment encompasses two regimens: drug-susceptible (DS) and -resistant (DR). DS-TB patients undergo a six-month treatment course comprising an intensive phase with rifampicin (RIF), isoniazid (INH), ethambutol (EMB), and pyrazinamide (PZA) for two months, followed by a four-month continuation phase with RIF and INH ^5^. For DR-TB cases, treatment regimens are adjusted based on resistance profiles, often requiring second-line drugs and extended treatment durations, which complicate patient management and result in poorer treatment outcomes (Seaworth, 2017).
Previous studies have shown that TB patient responses to standard anti-TB treatment vary depending on the infecting MTBC lineages, notably their immune response ^6, 7, 8, 9, 10^. Moreover, hypervirulent strains can dampen immune defences, leading to accelerated disease progression and increased transmission rates ^11^.
The mutation rates of lineages have been found to vary significantly, with some lineages exhibiting a higher propensity for developing mutations that confer resistance to primary TB drugs ^12^. For example, L2 has been noted for its increased mutation rates, particularly in drug-resistant (DR) strains, contributing to intrinsic treatment challenges and the emergence of multidrug-resistant (MDR) TB ^13, 14, 15^ The persistence and proliferation of resistant strains during treatment can lead to therapeutic failures and complicate TB control efforts ^16^.
This study investigates the genetic diversity of MTBC strains circulating in The Gambia over nearly two decades (2002 to 2021) and explores their implications for effective TB management. By integrating structural bioinformatics and computational approaches, we analysed the most extensive WGS collection of MTBC isolates in a West African country. We present the abundance, distribution and effects of genetic mutations on drug-target protein stability and conservation properties. Understanding these genetic variations is crucial for designing new drugs and developing effective TB treatment strategies, particularly in regions like West Africa, where multiple MTBC lineages coexist.
Methodology
Ethical statement
This study received ethical approval from the Medical Research Council Unit, The Gambia, at the London School of Hygiene and Tropical Medicine (MRCG@LSHTM)/The Gambian Government joint ethical committee. All recruited study participants or guardians provided written informed consent.
Sequencing and epidemiological data
The whole genome sequences (WGS) and epidemiological data used in this analysis (n=1803) were sourced from consecutive TB projects hosted by the TB case contact platform at the MRCG@LSHTM between 2002 and 2020. These projects include the following with their respective number of WGS samples: PRJEB53138 (Enhance Case Finding; n=1302), SCC 1289 (Childhood TB Program; n=234), SCC 1523 (TB Sequel; n=216) and Recurrent TB (n=52). The isolate’s metadata used in this study included age, sex and collection year.
Microbiology and DNA Extraction
Briefly, stored MTBC isolates from archived stocks or directly from a microbiology growth indicator (MGIT^™^) positive tube were subcultured into Middlebrook 7H9 broth or Lowenstein-Jensen (LJ) slopes to multiply the colonies. Genomic DNA was extracted using the cetyltrimethylammonium bromide (CTAB) method, as previously described ^17^. The extracted DNA underwent WGS on the Illumina HiSeqX platform at the Forschungszentrum Research Center Borstel, Germany. For the PRJEB53138 isolates, the DNA was extracted using Maxwell^®^ 16 Viral Total Nucleic Acid Purification Kit (Promega Corporation, Fitchburg, WI, USA) following the manufacturer’s instructions and sequenced in MicrobsNG in the United Kingdom.
Bioinformatic and phylogenetic analysis
Raw sequence data (approximately 2000 samples) was processed and analysed to ensure data quality and accuracy. The Kraken2 database tool was used to filter contaminated sequences and exclude non-MTBC strains. Poor-quality reads were trimmed using Trimmomatic (v0.39) with the following parameters: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:36. The quality of the processed reads were reassessed using FastQC.
For each sample, trimmed reads were aligned to the Mycobacterium tuberculosis H37Rv reference genome (accession: NC_000962.3) using BWA-MEM software ^18^. Single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) were identified through the application of the Genome Analysis Toolkit (GATK) ^19^ and Sequence Alignment Map (SAM) ^20^ tools. Genomic VCF files from all the samples were merged, and multi-FASTA alignments were generated using BEDTools software.
Phylogenetic relationships between the samples were inferred by constructing a phylogenetic tree with IQ-TREE software ^21^. The tree was visualised and annotated using the Interactive Tree of Life (iTOL) v6 software platform ^22^. The tree was visualised and annotated using the iTOL software platform. MTBC lineages and genotypic drug resistance profiles for each isolate were determined using the TB-Profiler pipeline (v4.4.0; database version: e25540b) ^23^. Variants, including missense and frameshift mutations within known drug resistance loci, were analysed and compared to established databases such as TB-Profiler and the WHO catalogue to identify reported and potential unreported polymorphisms. Mutations in Tier1 genes for The Gambian dataset were compared with global mutation data (>100K mutations) and specific datasets from other West African countries ^24, 25^.
The analysis included detailed information for each mutation, including the gene names, nucleotide changes, mutation frequency, and count for each country, identified by their country codes. This comprehensive approach provided insight into regional and global genetic diversity and drug resistance dynamics in MTBC strains.
Protein structural modelling and mutant stability prediction
Missense mutations associated with first-line drugs (RIF, INH, PZA, and EMB) were filtered using a frequency cutoff of 80% to ensure accuracy and relevance. Redundant mutations within the same gene were identified and removed to clarify the dataset and eliminate duplication. The final dataset comprised 943 isolates and their associated missense mutations, of which 614 were classified as susceptible and 329 as resistant. This curated dataset offers a comprehensive overview of the genetic basis of drug susceptibility and resistance.
Protein structures for the target genes were obtained from the Protein Data Bank (PDB), a key repository of experimentally determined macromolecular structures critical for biomedical research and drug discovery ^26, 27^. This dataset included four PDB files derived from experimentally determined crystal structures and twenty predicted structures using AlphaFold, a protein structure prediction tool ^28^. To predict the stability changes caused by mutations, the Delta Delta G (ΔΔG), representing the difference in free energy between wild-type and mutant protein forms, was calculated using PyRosetta, FoldX, and site-directed mutator (SDM) (https://compbio.medschl.cam.ac.uk/sdm2/) ^29, 30, 31^.
ConSurf (https://consurf.tau.ac.il/consurf_index.php) was employed to evaluate the evolutionary conservation of amino acids at mutation sites. Conservation grades, ranging from 1 (highly variable) to 9 (highly conserved), were assigned based on the evolutionary significance of specific residues ^32^. Highly conserved residues are often critical for structural integrity or functional roles in proteins. The conservation grades for mutation positions were extracted and compared between mutations classified as resistant and susceptible. This comparison offered insights into the mutations’ evolutionary importance and potential functional consequences.
Results
Study Demography
This study analysed 1803 MTBC isolates with whole-genome sequencing (WGS) data from TB patients residing in the Greater Banjul Area, which accounts for 80% of all TB cases in The Gambia. Metadata, including either age, sex, or year of sample collection, was available for 1713 isolates (95%) (Table 1). The majority of isolates (1145/1585, 72.2%) were from male patients, with the highest representation from individuals aged 18–29 (498, 32%) and 30–44 (409, 26.3%) years. Most samples were from patients diagnosed between 2012 and 2015 (1433/1713, 83.6%) (S1 figure).
MTBC lineages
Phylogenetic analysis revealed the clustering of isolates by lineage, confirming the dominance of specific MTBC lineages in the population (Figure 1). Most isolates (94%) belonged to Mb Lineage 4 (L4; 1214/1804, 67.2%) and Maf Lineage 6 (L6; 480/1804, 26.6%) (Table 1). L4 has remained the predominant lineage throughout the study period (S1 figure). Among the sub-lineages, L4.1 (410 isolates) and L4.3 (LAM; 323 isolates) were the most abundant within L4, while L6.1 was the most common sub-lineage of L6 (S2 figure).
Drug resistance
The genotypic resistance profile revealed that 1421 (78%) were drug-susceptible (DS). Among the drug-resistant (DR) isolates, 90 (5.0%) were resistant to INH alone, 10 (0.6%) were resistant to RIF alone, and 19 (1.1%) were multi-drug resistant (MDR). Additionally, 282 isolates (15.3%) were classified as other, having potential drug-resistance mutations, identified by the TB-Profiler pipeline ^23^. The most frequent mutation underlying resistance to INH was katGSer315Thr, observed in 71 isolates (71/90, 78.8%) (Table 2). Lineage-specific mutations were also detected in known drug-resistance genes. For instance, embC Ala307Thr, a mutation associated with ethambutol but classified as uncertain significant by the WHO catalogue, was specific to L6, with a frequency of 56.34% (270/480). Another ethambutol-associated uncertain significant mutation, embA Thr113Arg, was also unique to L6 with a frequency of 22.61% (106/480) (Table 2). According to the drug resistance classification, INH resistance (HR-TB) and MDR-TB were predominantly observed in lineage 4. In contrast, lineage 6 exhibited the highest number of resistances classified as “Other,” including mutations of uncertain significance defined by the WHO catalogue. INH and these “Other” resistances were mainly detected between 2012 and 2019 (S3 figure).
Genetic variability
To investigate the specificity of genetic mutations in MTBC isolates from The Gambia, we compared missense mutation frequencies in Gambian isolates to those in the rest of West Africa and global datasets. Among over 100,000 mutations identified across 100 countries, 12,411 mutations in all drug resistance genes (11.6%) originated from The Gambia, underscoring the country’s notable contribution to the global mutation pool. Mutations in genes associated with drug resistance were found at significantly higher frequencies in The Gambia compared to global averages, particularly in key genes such as rpoB (RIF resistance), inhA (INH resistance), and embB (EMB resistance). For example, the rpoB Thr350Ile uncertain significant mutation by the WHO catalogue was observed in 26.9% of Gambian isolates compared to 0.99% globally (Figure 2A). The embC Ala307Thr uncertain significant mutation appeared in 15.7% of Gambian isolates but was rare globally (Figure 2C). These mutations were steadily identified in MTBC isolates over the study period (S4 figure). In contrast, uncertain significant mutations associated with resistance to second-line drugs, such as moxifloxacin, were more common in The Gambia and West Africa than the global average. For example, gyrB Ala403Sep and gyrA Leu398Phe were both found at higher frequencies in The Gambia (34% and 29%) and West Africa (41% and 31%), respectively, compared to the global frequency (S5 figure). Some non-resistance mutations, like c.−100C>T, were uniquely prominent in The Gambia, often co-occurring with ethambutol-resistance (S6 figure).
Structural and conservation properties of resistance and susceptible mutations
The distributions and structural properties of mutant sites were analysed based on residue depth, occluded surface packing (OSP), and relative solvent accessibility (RSA). A significant difference was observed between resistance and susceptible mutations in all categories of the structural properties (two-tailed Mann-Whitney test, p<0.05). It is interesting to note that the resistance and susceptible mutations occur more frequently in tightly (OSP > 0.4) and less tightly packed (OSP < 0.4) environments in the protein structure, respectively (Figure 3A). In terms of residue depth, susceptible mutations were observed more often at shallow residue depths (< 4 Å), while resistant mutations were concentrated at greater depths (8–9 Å) (Figure 3B). Similar trends were also observed for RSA, where the resistance mutations were found at higher frequency in solvent-inaccessible regions (RSA < 20%). In contrast, susceptible mutations were more frequently located in solvent-accessible regions (Figure 3C).
Additionally, the conservation levels of resistance and susceptible mutations were analysed. Resistance mutations exhibited a significantly higher conservation level than background and susceptible mutations. Specifically, at the conservation grade from 5 to 9, the density of resistant mutations surpassed that of background and susceptible mutations. Conversely, at conservation grades of 1 to 4, the density of susceptible mutations was more prevalent than background and resistant mutations (Figure 3D). Overall, resistant mutations displayed more significant conservation than susceptible mutations (one-tailed Wilcoxon matched-pair signed rank test, p<0.05), highlighting their potential functional and evolutionary significance.
Mutant stability prediction
The values of the ΔΔG (change in free energy) were analysed to predict the impact of resistant and susceptible mutations on protein stability. No significant differences in ΔΔG were observed between resistance and susceptible mutation using FoldX and SDM (Figure 4). SDM predicted median ΔΔG values of −0.21 for resistance and −0.18 for susceptible mutations. On the other hand, FoldX predicted median ΔΔG values of 0.31 for resistance and 0.24 for susceptible mutations. Both tools suggested that these mutations could severely destabilise or stabilise the protein (absolute ΔΔG > 0.5).
In contrast, PyRosetta analysis revealed significant differences in ΔΔG values (two-tailed Mann-Whitney test, p<0.05). Resistant mutations showed a relatively more significant destabilising effect (median ΔΔG=−52.74) compared to susceptible mutations (median ΔΔG=−62.28) (Figure 4).
To further investigate ΔΔG differences between resistant and susceptible mutations, mutations were grouped by first-line anti-tuberculosis drugs (RIF, INH, EMB, and PZA) and categorised by MTBC lineages. No significant differences between resistant and susceptible mutations were observed for any of the four drugs using SDM (S7 figure). In contrast, PyRosetta analysis revealed statistically significant differences (two-tailed Mann-Whitney test) for INH in Lineage 6 and EMB in Lineage 4 (Figure 5A). FoldX analysis also identified substantial differences in ΔΔG values for RIF in Lineage 6 and EMB and PZA in Lineage 2 (Figure 5B).
Discussion
This study provides insights into the genomic landscape of Mycobacterium tuberculosis complex (MTBC) isolates in The Gambia for nearly two decades, shedding light on the country’s epidemiology, lineage diversity and drug resistance (DR) patterns. Consistent with prior studies in West Africa (De Jong et al., 2010), MTBC Lineage 4 (L4) and 6 (L6) were found to dominate, collectively accounting for 94% of TB cases.
L4, the most prevalent lineage globally, represented 67.2% of isolates, likely due to its high virulence ^33^, rapid progression to active TB ^34^, and adaptability to diverse environments ^35^. Within L4, the overrepresentation of sub-lineages L4.3 (LAM) and L4.1 further supports the role of these genetic clusters in driving its success in this population. LAM is a well-known lineage associated with increased virulence and transmission potential, which may explain its widespread distribution in this region ^36^
In contrast, L6, which accounted for 26.6% of the isolates, remains mainly geographically restricted to West Africa. This highlights the importance of region-specific interventions considering the local strains’ genetic makeup ^37^. Despite its limited global prevalence, this study’s significant presence of L6 aligns with its known confinement to the region. The predominance of sub-lineage L6.1 highlights its evolutionary adaptation and persistence within this population, likely shaped by unique host-pathogen interactions in the region ^38^.
The demographic trends observed are consistent with global TB patterns. Most cases were identified in male patients (72.5%), reflecting the higher incidence of TB among men worldwide ^1^. The patient’s age distribution, with the highest burden among individuals aged 18–29 and 30–44, underscores the significant impact of TB on economically and socially critical population segments in The Gambia. These findings emphasise the importance of targeted interventions to mitigate the disease’s socioeconomic impact in The Gambia.
The detection of 19 multidrug-resistant (MDR) isolates highlights the pressing challenge of TB in The Gambia, given the limited treatment options and increased risk of treatment failure associated with mistreated MDR TB. The higher prevalence of isoniazid (INH) mono-resistance (90isolates) comparedto rifampicin (RIF) mono-resistance (10 isolates) suggests that INH resistance may be an emerging issue in The Gambia. This finding underscores the need for continuous surveillance and updated treatment protocols that reflect the evolving resistance patterns. Furthermore, the study’s reliance on genotypic data to infer DR highlights the importance of integrating molecular diagnostics into routine TB control programs, which can facilitate the timely and accurate detection of DR strains. The high frequency of specific mutations, such as rpoB Thr350Ile and embC Ala307Thr (EMB), highlights the importance of region-specific genomic surveillance to guide TB control efforts effectively. Identifying mutations with uncertain significance in the WHO catalogue, particularly those with higher frequencies in The Gambia, highlights a critical knowledge gap. Investigating these mutations’ clinical relevance, fitness effects, and contribution to DR through in vitro and in vivo studies will enhance our understanding of DR mechanisms.
Analysis of structural properties revealed key differences between resistant and susceptible mutations. Resistant mutations were more frequently located in tightly packed regions of the protein structure (OSP>0.4) and solvent-inaccessible regions (RSA < 20%), suggesting that they may disrupt proteins’ core structural stability ^39^. In contrast, susceptible mutations were more often in less tightly packed, solvent-accessible regions. Additionally, resistance mutations exhibited higher conservation grades (5–9) than susceptible and background mutations, indicating their critical role in maintaining essential protein functions. These findings align with the hypothesis that resistance mutations often occur at functionally or structurally critical residues under strong evolutionary constraints ^40^. Understanding these distinctions could inform drug design by prioritising highly conserved and structurally significant target sites.
The stability analysis demonstrated variations in the predictive capabilities of different computational tools. While SDM and FoldX did not identify significant differences in ΔΔG values between resistant and susceptible mutations, PyRosetta revealed substantial differences. Resistant mutations showed a relatively more destabilising effect (median ΔΔG= −52.74) compared to susceptible mutations (median ΔΔG= −62.28), suggesting that PyRosetta may be more sensitive. These destabilising effects may reflect structural alterations that disrupt drug-binding interactions or enable conformational changes, thereby reducing drug efficacy.
Grouping mutations by first-line anti-TB drugs and MTBC lineages provided further resolution. For example, PyRosetta detected significant ΔΔG differences for NIH mutations in lineage 6 and EMB for Lineage 4, while FoldX highlighted significant differences for RIF (Lineage 6) and EMB and PZA (Lineage 2). These lineage-specific findings underscore the importance of considering genetic background and selective pressures when studying resistance mechanisms and designing treatment strategies.
While this study offers valuable insights into the genomic landscape of MTBC isolates in The Gambia, several limitations must be acknowledged. First, there is a potential underestimation of lineage 6 prevalence, as genotyping was primarily conducted from subcultured isolates, which may introduce culture bias against the slow-growing L6 isolation. Future studies could consider directly genotyping from sputum samples, minimising the culture bias associated with subculturing from freezer-stored isolates. Second, the functional significance of many identified mutations, particularly those classified as having “uncertain significance,” requires further investigation to clarify their clinical relevance. Experimental validation through in vitro and in vivo studies is essential. Thirdly, while tools like AlphaFold provided valuable insights, their accuracy in predicting the effect of mutations on protein stability (ΔΔG) remains limited ^41^. Integrating multiple predictive methods with experimental validation will enhance our understanding of mutation impacts. Lastly, only some of the WGS isolates had phenotypic resistance data, which limits our ability to confirm these mutations’ importance in drug resistance pathways.
Despite these limitations, this study highlights the importance of lineage-specific mutations providing a robust framework for future research. Addressing these gaps will improve our understanding of the genomic and structural mechanisms underlying DR and inform more effective TB control strategies.
Conclusion
Our genome-wide analysis of Mycobacterium tuberculosis complex (MTBC) strains in The Gambia reveals a diverse landscape of circulating lineages and drug resistance profiles. L4 and L6 dominate the region, with L4 exhibiting global adaptability and virulence, while L6 remains regionally confined to West Africa. The coexistence of global and local lineages underscores the need for tailored, region-specific TB control strategies.
Drug resistance mutations, such as katG Ser315Thr and embC Ala307Thr, were notably frequent. Lineage-specific variants like embC Ala307Thr in L6 warrant further investigation for their roles in ethambutol resistance and bacterial fitness. Structural analyses highlighted that resistance mutations often occur in solvent-inaccessible, highly conserved regions, impacting protein stability and evolutionary fitness. These findings underscore the complexity of resistance mechanisms and their implications for treatment outcomes.
Finally, our study emphasises the importance of integrating genomic surveillance with functional validation to bridge the gap between genomic data and clinical outcomes. By refining our understanding of MTBC evolution, epidemiology, and resistance mechanisms, these efforts will inform tailored strategies to combat TB in The Gambia and contribute to global efforts to end TB.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health, O. Global tuberculosis report 2023. World Health Organization: Geneva, 2023.
- 2Ngabonziza J.C.S. A sister lineage of the Mycobacterium tuberculosis complex discovered in the African Great Lakes region. Nat Commun 11,2917 (2020).32518235 10.1038/s 41467-020-16626-6PMC 7283319 · doi ↗ · pubmed ↗
- 3Napier G. Robust barcoding and identification of Mycobacterium tuberculosis lineages for epidemiological and clinical studies. Genome medicine 12,1–10 (2020).10.1186/s 13073-020-00817-3PMC 773480733317631 · doi ↗ · pubmed ↗
- 4De Jong B.C., Antonio M. & Gagneux S. Mycobacterium africanum—review of an important cause of human tuberculosis in West Africa. P Lo S neglected tropical diseases 4,e 744 (2010).20927191 10.1371/journal.pntd.0000744 PMC 2946903 · doi ↗ · pubmed ↗
- 5Fattorini L. & Iacobino A. Drug Resistance Mechanisms In Tuberculosis | Encyclopedia. 2020; 2020.
- 6Diarra B. Mycobacterium africanum (Lineage 6) shows slower sputum smear conversion on tuberculosis treatment than Mycobacterium tuberculosis (Lineage 4) in Bamako, Mali. P Lo S One 13,e 0208603 (2018).10.1371/journal.pone.0208603 PMC 629112430540823 · doi ↗ · pubmed ↗
- 7Tientcheu L. Host Immune Responses Differ between M. africanum- and M. tuberculosis-Infected Patients following Standard Anti-tuberculosis Treatment. PLOS Neglected Tropical Diseases 10,e 0004701 (2016).10.1371/journal.pntd.0004701 PMC 487158127192147 · doi ↗ · pubmed ↗
- 8Click E.S., Winston C.A., Oeltmann J.E., Moonan P.K. & Mac Kenzie W.R. Association between Mycobacterium tuberculosis lineage and time to sputum culture conversion. Int J Tuberc Lung Dis 17,878–884 (2013).23743308 10.5588/ijtld.12.0732 · doi ↗ · pubmed ↗