Presentation, Treatment, and Outcome of a First Stroke-like Episode in a Carrier of the Compound Heterozygous Variants c.695G_A and c.2209G_C in POLG: A Case Report
Dominik Zieglgänsberger, Josef Finsterer

TL;DR
A 19-year-old woman with POLG mutations experienced a first stroke-like episode and seizures, highlighting the complex clinical presentation and treatment challenges of this genetic condition.
Contribution
This is the first reported case of POLG-related polyneuropathy progressing to a stroke-like episode and seizures in a carrier of compound heterozygous POLG variants.
Findings
The patient's POLG mutations initially presented as polyneuropathy and later as a stroke-like episode with seizures.
Treatment of status epilepticus required thiopental anesthesia, while levetiracetam, lacosamide, and perampanel showed long-term efficacy.
The stroke-like lesion initially worsened before regressing under treatment with glucocorticoids, NO precursors, and antioxidants.
Abstract
In this report, we describe the case of a 19-year-old female patient with two heterozygous POLG variants in different exons manifesting phenotypically as polyneuropathy and later with a first stroke-like episode (SLE) and seizures. This has not been reported earlier. The patient suffered from POLG-related neuropathy since the age of 14 and was admitted for her first SLE, which manifested clinically as hemianopia and two days later led to impaired consciousness, hemiparesis, seizures, and status epilepticus. Cerebral MRI showed a corresponding stroke-like lesion (SLL) in the right occipital lobe. The seizures were treated with levetiracetam, lacosamide, and perampanel, but the treatment of status epilepticus required anesthesia with thiopental, ketamine, and midazolam. In addition, glucocorticoids, nitric oxide (NO) precursors, and antioxidants were administered. Under this treatment,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
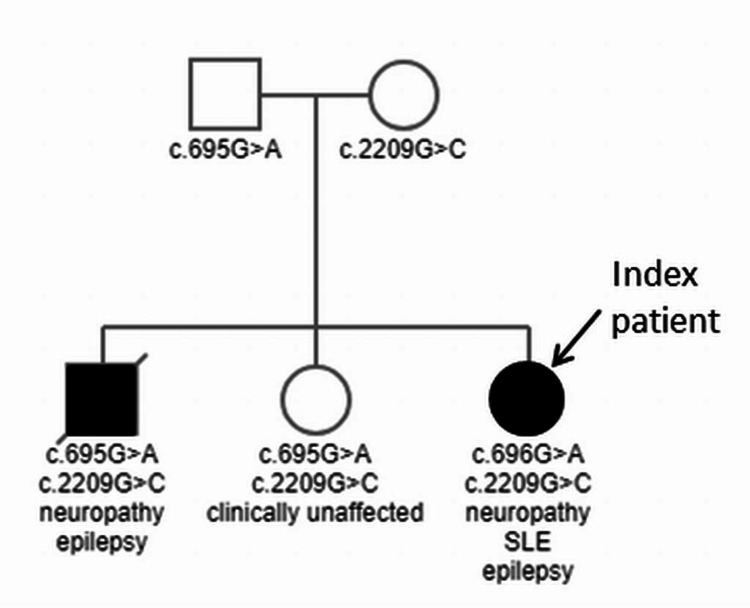 Figure 1
Figure 1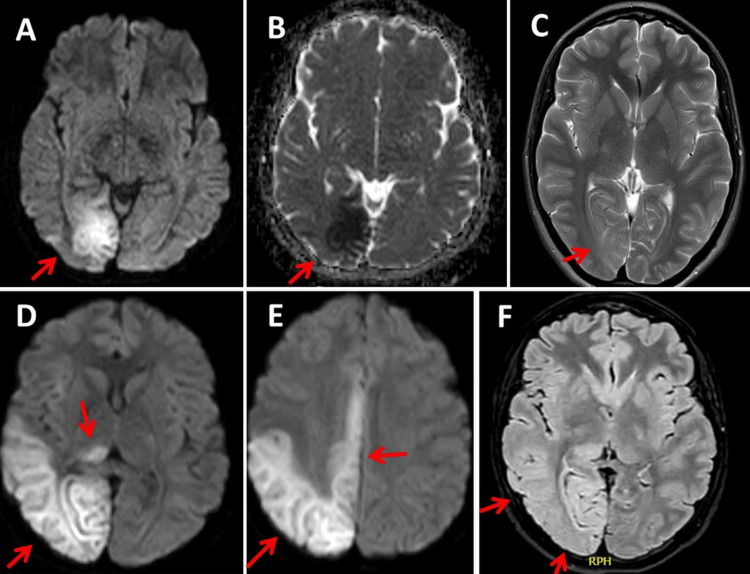 Figure 2
Figure 2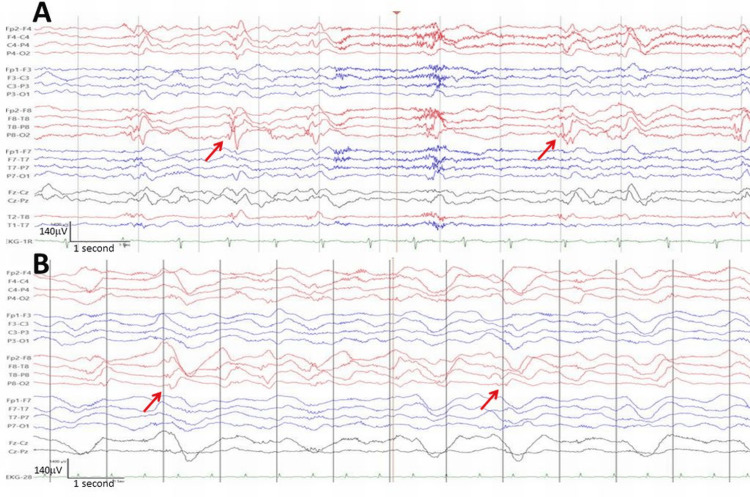 Figure 3
Figure 3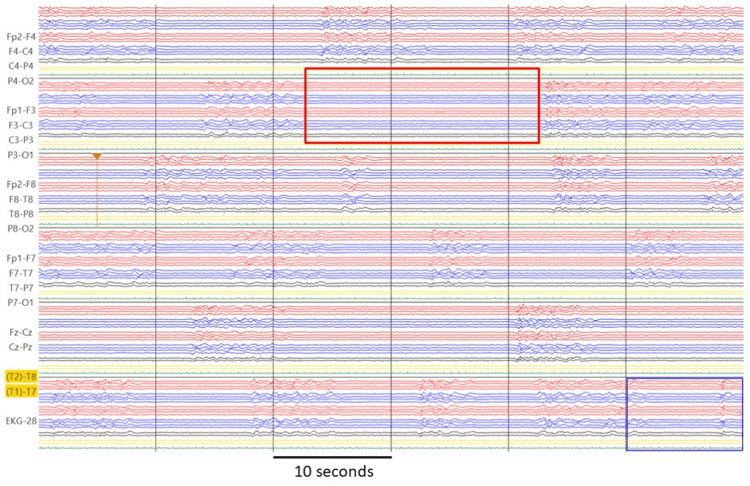 Figure 4
Figure 4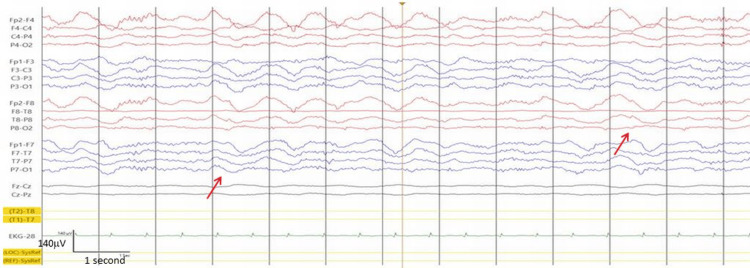 Figure 5
Figure 5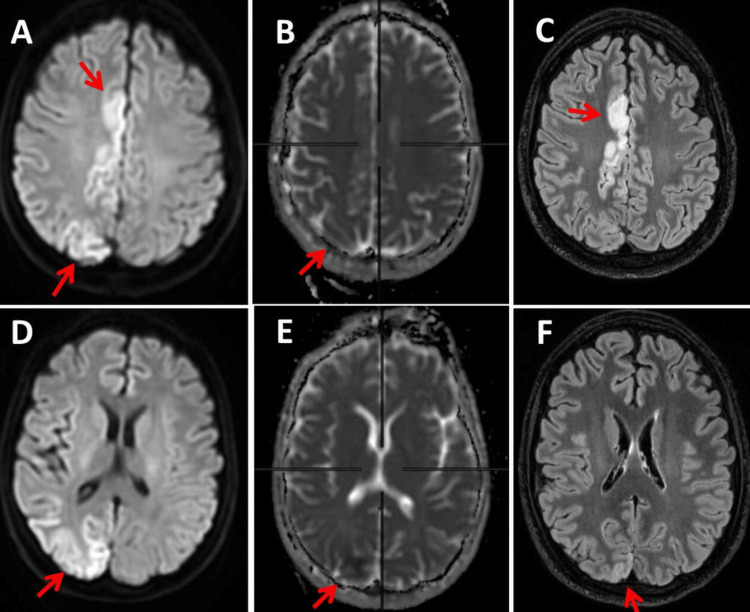 Figure 6
Figure 6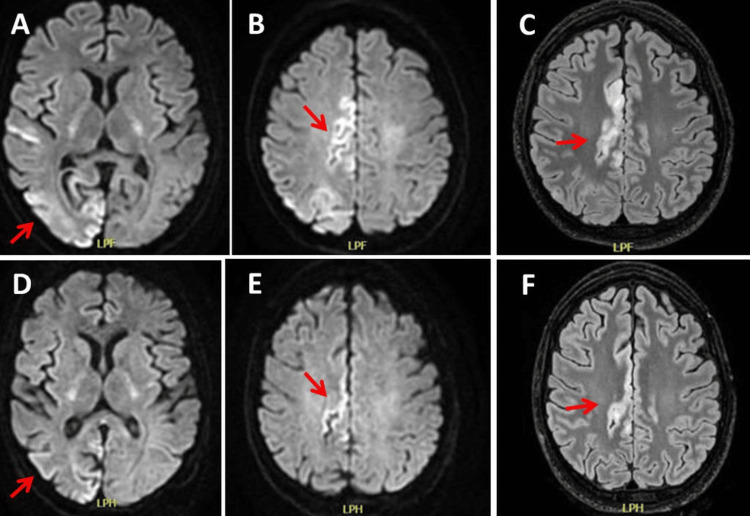 Figure 7
Figure 7| Age (years) | Hospital Day | Diagnosis/event | Treatment |
| 14 | axonal neuropathy | wheelchair for long distances | |
| 19 | 1 | SLE (hemianopia), epileptiform discharges | LEV |
| 19 | 3 | hemiparesis, status epilepticus | LEV, LCM, PER |
| 19 | 4 | status epilepticus | midazolam |
| 19 | 5 | status epilepticus | midazolam increased |
| 19 | 6 | status epilepticus, BSP | thiopental, ketamine |
| 19 | 7 | BSP | thiopental increased |
| 19 | 8 | BSP | thiopental reduced |
| 19 | 9 | BSP | thiopental reduced |
| 19 | 10 | BSP | thiopental reduced |
| 19 | 12 | BSP, rhythmic delta | thiopental withdrawn |
| 19 | 13 | regression of SLL | ketamine reduced |
| 19 | 14 | none | ketamine reduced |
| 19 | 15 | spiky EEG | ketamine reduced |
| 19 | 17 | none | ketamine withdrawn |
| 19 | 18 | none | midazolam reduced |
| 19 | 19 | none | midazolam reduced |
| 19 | 21 | none | midazolam withdrawn |
| 19 | 27 | further regression of SLL | none |
| 19 | 28 | tracheotomy, recurrent focal status | midazolam |
| 19 | 52 | further regression of SLL | none |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMitochondrial Function and Pathology · Cardiovascular Function and Risk Factors · Advanced MRI Techniques and Applications
Introduction
Mitochondrial disorders (MIDs) are not only caused by mutations in mtDNA but also by those in nuclear DNA. One of the nuclear genes that are frequently mutated in MIDs is POLG. POLG variants present with heterogeneous phenotypes known as POLG spectrum disorders, ranging in severity from asymptomatic or mild to fatal [1]. A rare clinical manifestation of POLG variants is hereditary neuropathy, which often manifests together with dysarthria/dysphagia and ophthalmoplegia (sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) syndrome) [2]. Much more frequently than in the peripheral nervous system (PNS), POLG variants manifest phenotypically in the central nervous system (CNS) [3]. POLG phenotypes with CNS manifestations include Leigh syndrome, MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) syndrome, Alpers-Huttenlocher syndrome (AHS), also known as progressive sclerosing poliodystrophy, and MNGIE (mitochondrial neuro-gastro-intestinal encephalopathy) syndrome, mitochondrial depletion syndrome (MDS), mitochondrial recessive ataxia syndrome (MIRAS), spinocerebellar ataxia with epilepsy (SCAE), myoclonic epilepsy, myopathy and spinocerebellar ataxia (MEMSA), isolated epilepsy, Parkinson's syndrome, dementia, developmental delay, and rare stroke-like episodes (SLEs) [3-6]. POLG-related disease is diagnosed based on clinical presentation, cerebral imaging, EEG, and evidence of a pathogenic POLG variant.
The POLG gene is located on the long arm of chromosome 15 in position 26.1 and has 23 exons. It produces a 140 kDa protein consisting of 1239 amino acids [7]. The protein encoded by POLG belongs to the family of type A DNA polymerases. It is a mitochondrial nucleoid with a Mg2+ cofactor and 15 turns, 52 beta-strands, and 39 alpha-helixes [7]. POLG contains a polyglutamine tract near its N-terminus, which may be polymorphic. Two transcript variants encoding the same protein have been found for this gene: POLG1 and POLG2 [7]. POLG is the only known DNA polymerase in human mitochondria and is essential for mitochondrial DNA replication and repair [8]. It is known that defects in mtDNA replication lead to mitochondrial dysfunction and disease. Several hundred coding variations have been identified in the gene encoding the catalytic subunit of POLG [8]. However, a patient carrying two heterozygous POLG variants in different exons, phenotypically manifesting initially as polyneuropathy and later with an SLE and seizures, has not yet been described.
Case presentation
The index patient was a 19-year-old woman with POLG-related MID due to c.695G>A and c.2209G>C variants, who initially manifested with neuropathy at the age of 14 years. The neuropathy, which manifested clinically with sensory disturbances and exercise intolerance, was classified as axonal sensorimotor polyneuropathy based on nerve conduction studies and required the use of a wheelchair for long distances (Table 1). At 19 years of age, the patient had the first SLE with the initial clinical manifestation being hemianopia to the left on waking in the morning, which led to hospitalization (hospital day (HD) 1).
The patient's medical history was negative for epilepsy. Family history was positive for a POLG-related disorder in the brother, who died of complications after prolonged status epilepticus at the age of 20 (Figure 1). In addition to epilepsy, he had shown phenotypic polyneuropathy from the age of 13, which is why a genetic investigation was initiated involving his parents and sisters, in which the variants c.695G>A in exon 3 and c.2209G>C in exon 13 of the POLG gene were detected in all three siblings (Figure 1). The c.695G>A variant was inherited from the father and the c.2209G>C variant from the mother (Figure 1). The c.695G>A variant resulted in the amino acid change p.Arg232His, rs113994093, and the c.2209G>C variant resulted in the amino acid change p.Gly737Arg. s121918054. The mother also carried the variant c.2171G>A (p.Gly724Glu, rs201524659) in exon 14 of the TT-K2 gene and the father also carried the heterozygous variant c.2248G>A (p.Val750lle, rs148171058) in exon 14 of the TRPV4-0 gene and the variant c.1550G>T (p. Gly517Val, rs61752783) in exon 8 of the POLG gene. The index patient’s 16-year-old sister also carried the two pathogenic POLG variants but was not yet phenotypically apparent.
Pedigree with transmission of the variant c.695G>A from the mother and the variant c.2209G>C from the father to all three siblings, of whom only the index patient and her brother were clinically affected
MRI of the brain on admission showed an occipital lesion that was hyperintense on T2/fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted imaging (DWI) and hypointense on apparent diffusion coefficient (ADC) (Figure 2). EEG showed focal slowing in the right posterior region with interictal epileptiform discharges. Treatment with levetiracetam (LEV) 2000 mg/day was started.
MRI of the brain at admission (Day 1) shows an occipital lesion that was hyperintense on T2/FLAIR (panel C) and DWI (panel A) and hypointense on ADC (panel B). The cerebral MRI on hd3 shows an enlargement of the right occipital lesion (panels D, E, F)FLAIR: fluid-attenuated inversion recovery; DWI: diffusion-weighted imaging; ADC: apparent diffusion coefficient
On HD 3, impaired consciousness and left-sided hemiparesis occurred. MRI showed an enlargement of the right occipital lesion (Figure 2). EEG showed lateral periodic discharges (LPDs) or status epilepticus (Figure 3). Antiseizure medication (ASM) was increased to LEV 3000 mg/day, lacosamide (LCM) 400 mg/day, and perampanel (PER) 12 mg/day. In addition to the ASM, the patient received a single dose of L-arginine (0.5 g/kg), which has since been continued at 0.1 g/kg.
EEG recorded on hospital day 3 with LPDs or status epilepticus (panel A). LPDs were also recorded on hospital day 6 (panel B) (horizontal bar: 1s, vertical bar: 140V)LPD: lateral periodic discharge;
On HD 4, the EEG still showed LPDs, so ASM was extended to LCM 400 mg/day, PER 12 mg/day, and midazolam 45 mg/hour. Midazolam was increased to 60 mg/hour on HD 5. As the EEG was more or less unchanged on HD 6 (Figure 3), ASM was extended to intubation and anesthesia with thiopental (250 mg/hour) and ketamine (200 mg/hour), resulting in a burst suppression pattern (BSP) with burst durations of initially five to eight seconds and suppression of up to 20 seconds (Figure 4). In addition, methylprednisolone (1000 mg/day) was administered over four days (HD 6-9). On HD 7, suppression of up to 17 minutes was achieved with thiopental 250 mg/hour. On HD 8, thiopental was reduced to 200 mg/hour, on HD 9 to 150bmg/hour, and on HD 10 to 100 mg/hour. Riboflavin and coenzyme-Q were added. After discontinuation of thiopental on HD 12, continuous rhythmic delta activity was observed (Figure 5). MRI scan on HD 13 showed regression of the occipital lesion but a new DWI-hyperdense lesion in the right thalamus and cortical band, which is common in status epilepticus. Ketamine was reduced from 150 to 100 mg on HD 14 and 75 mg on HD 15 and discontinued on HD 17. Despite the reduction in anesthetics, the EEG still showed spikes on HD 15. Midazolam was reduced to 40 mg/hour on HD 18 and to 20 mg on HD 19 and discontinued on HD 21. The cerebral MRI on HD 27 showed further regression of the right occipital lesion (Figure 6).
Monitor EEG recording on hospital day 7 under thiopental 250 mg/hour with BSP, with suppression up to 20 seconds (horizontal bar: 10s)BSP: burst suppression pattern
Routine EEG recording on hospital day 13 with delta activity after termination of status epilepticus (horizontal bar: 1s, vertical bar: 140V)
Cerebral MRI on hospital day 27, one day before extubation with regression of the SLL on DWI (panels A, D), ADC (panels B, E), and FLAIR (panels C, F)SLL: stroke-like lesion; DWI: diffusion-weighted imaging; ADC: apparent diffusion coefficient; FLAIR: fluid-attenuated inversion recovery
After extubation on HD 28 and tracheostomy, the patient developed recurrent focal motor status epilepticus with clonus of the right face and right shoulder, which could be stopped by the renewed administration of midazolam (1 mg/hour) and boluses. Nevertheless, the cerebral MRI on HD 31 showed a further regression of the T2/FLAIR, DWI, and ADC lesion in the right occipital lobe. Almost complete resolution of the SLL was observed on HD 52 (Figure 7). She is currently on medication with LEV 3000 mg/day, LCM 400 mg/day, PER 12mg/day, L-arginine 15g/day, vitamin Q10 (111.5 mg/day, vitamin B1 2100 mg/day, folinic acid 50mg/day, and fentanyl 75 mcg/hour.
MRI of the brain on hospital day 27 (panels A-C) and on hospital day 52, showing further resolution of the SLL on T2 (panels D, E) and FLAIR (panel F)SLL: stroke-like lesion; FLAIR: fluid-attenuated inversion recovery
Discussion
This case is interesting in several respects. First, the patient carried a novel POLG variant in exon 3, which was found in compound heterozygosity with a rarely described POLG variant in exon 13. Second, the first clinical manifestation of the variant was an axonal sensorimotor neuropathy in the index patient and her brother. Third, the index patient suffered from SLE, which has rarely been reported in POLG variants. Fourth, the generalized status epilepticus could only be suppressed by anesthesia with thiopental, ketamine, and midazolam. Fifth, ASM possibly together with NO precursors and antioxidants had a favorable effect not only on seizure activity but also on SLL dynamics as an imaging correlate of SLE.
The variant c.2209G>C, which leads to the amino acid change p.Gly737Arg, has been reported previously by Phillips et al. [9], and classified as pathogenic or probably pathogenic [10]. In Phillips et al.'s case, their 20-year-old female patient showed slowed psychomotor development, delayed walking at three years of age, and delayed speech [9]. She subsequently developed progressive balance problems, spasticity, and pes equinus requiring Achilles tendon lengthening, tibialis posterior tendon transfer, the Jones procedure, and a walking aid from the age of 15. From that time, she also had achalasia with vomiting that required dilatations. At the age of 16, their patient presented with severe weakness of the ankle dorsiflexion, weakness of the intrinsic hand muscles, and moderate weakness of the hamstring muscles. There was pinprick hypoesthesia and pallhypoesthesia in the distal lower limbs. She was areflexive in the arms and legs. Nerve conduction studies (NCS) revealed severe axonal sensorimotor polyneuropathy. Their patient also carried two heterozygous variants in POLG, the variant c.2209G>C in exon 13 and the variant c.926G>A in exon 4 of POLG [9]. However, in contrast to the index patient, the patient reported by Phillips et al. did not manifest in the CNS.
The c.2209G>C variant was also reported in another patient, a 30-year-old man who presented phenotypically with tremors, dystonia, bradykinesia, and sensory disturbances since the age of 23 [11]. The tremor was associated with involuntary inversion of the feet and flexion of the right thumb, head posturing, and paresthesias in the right foot. Dystonia and tremor did not occur in the supine position, and there was a diurnal component that occurred only a few hours after waking. The patient was unable to walk in tandem but was able to walk on his heels and toes and support himself with his hand to maintain balance. NCS and somatosensory-evoked potentials (SSEPs) revealed evidence of moderate to severe generalized sensory length-dependent axonal polyneuropathy; cerebral and spinal MRI was not conclusive. A trial of low-dose levodopa/benserazide led to a significant improvement in tremor and dystonia; however, four years later, the patient also developed an onset of progressive external ophthalmoplegia, resting tremor in the right upper limb, occasional myoclonic jerks in the shoulder, arm, forearm, and distal fingers, appendicular cerebellar dysfunction, and mild bilateral foot drop. In addition to c.2209G>C, the patient carried the heterozygous variant c.3305A>C [11].
The reason why the phenotypic expression was heterogeneous in the index patient in the current report and the other two patients [9,11] remains speculative, but one reason could be that the second mutation besides the c.2209G>C variant was different in all three patients. The similarities between the three patients concerned the development of axonal polyneuropathy in all of them. However, the patient reported by Phillips et al. did not develop CNS manifestations [9], and the patient reported by Qiu et al. did not have SLEs [11]. However, SLE has also been reported in patients carrying variants other than those of the index patient. Cheldi et al. reported a 74-year-old man with SLE and myopathy due to the three heterozygous POLG variants c.752C>T in exon 3, c.1760C>T in exon 10, and c.3556G>C in exon 22 [12]. Surprisingly, the cerebral lesions found on imaging were interpreted as ischemic, but are more likely metabolic in nature given the multisystem nature of the disease and the genetic background. Martikainen et al. also reported SLE in a 26-year-old woman with headache, visual flashes, dysarthria, and generalized seizures [13]. EEG showed non-convulsive status epilepticus and cerebral MRI showed T2 hyperintensity in a parieto-occipital distribution consistent with SLL. Genetic testing revealed the c.2243G>C variant in POLG, and their patient benefited from phenytoin, oxcarbazepine, and LEV [13]. SLEs have also been reported in patients with AHS [14]. In a study of six POLG mutation carriers, all of them also had SLEs [15].
Seizures and status epilepticus are common manifestations of *POLG *variants. In a study of nine AHS patients by van Westrhenen et al., EEG recordings showed rhythmic high-amplitude deltas with superimposed (poly)spikes (RHADS) in all of them [16]. In six of the AHS patients, RHADS were present at first status epilepticus. Sometimes they appeared 5-10 weeks later and disappeared over time. RHADS were symptomatic in three AHS patients, and five AHS patients showed distinct ripples on the (poly)spikes of RHADS. In another study of five AHS patients by Wolf et al. with focal-clonic or complex-focal status epilepticus as the initial manifestation of the disease, three of them died 3-12 months after the onset of seizures [17]. In four children, the first EEG showed RHADS. Some of these patients did not respond adequately to established ASM and required more unusual therapy. In two women with AHS who manifested with seizures, these did not stop despite the use of multiple ASMs [18]. However, after the initiation of intravenous magnesium as ASM, clinical and neurophysiological improvement and rapid extubation of both patients were achieved. In general, there is no difference in the treatment of status epilepticus with and without SLE.
The pathophysiological consequences of POLG mutations are not always clear, but it is known that POLG mutations impair mtDNA synthesis and consequently lead to depletion and impaired repopulation of mtDNA in intact cells. A known pathophysiological consequence of POLG mutations is the upregulation of Notch and Janus Kinase-Signal Transducers and Activators of Transcription (JAK-STAT) signaling pathways. These signaling pathways are crucial for neuronal development. The upregulation of Notch signaling pathways also leads to stem cell activation and increases oxidative stress. Oxidative stress can be exacerbated by increased NO production due to endothelial and vascular dysfunction in cells with POLG mutations. Increased oxidative stress and increased NO production may be the reason why antioxidants and NO precursors are beneficial in patients with MID. Whether the use of NO precursors and antioxidants actually had a beneficial effect not only on seizure activity but also on the manifestations of SLL in the index patient remains speculative; however, since such a beneficial effect has been previously reported [19,20], it is conceivable that these drugs also favorably influenced the dynamics and manifestations of SLL. Regardless of these potential mechanisms and treatment effects, the most effective treatment for SLLs remains ASM when SLLs present with seizures.
The limitations of this case study were that biochemical assays were not performed, mtDNA copy number was not measured, and mtDNA was not sequenced to assess whether the POLG mutations caused secondary single or multiple mtDNA deletions.
Conclusions
This case demonstrates that pathogenic POLG variants may initially manifest phenotypically with polyneuropathy, POLG variants may manifest with SLE, which occurs before the onset of seizures, status epilepticus due to POLG variants may require thiopental anesthesia, and LEV, LCM, and PER may be beneficial for seizure activity in these patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A perspective on human cell models for POLG-spectrum disorders: advantages and disadvantages of CRISPR-Cas-based vs. patient-derived i PSC models Med Genet Cakmak C Zempel H 2452493320213883570310.1515/medgen-2021-2090 PMC 11006303 · doi ↗ · pubmed ↗
- 2Sensory ataxic neuropathy with dysarthria/dysphagia and ophthalmoplegia (SANDO). Two case reports Acta Myol Gáti I Danielsson O Jonasson J Landtblom AM 188190302011 https://pubmed.ncbi.nlm.nih.gov/22616202/22616202 PMC 3298093 · pubmed ↗
- 3POLG-related disorders and their neurological manifestations Nat Rev Neurol Rahman S Copeland WC 40521520193045197110.1038/s 41582-018-0101-0PMC 8796686 · doi ↗ · pubmed ↗
- 4POLG 1-related epilepsy: review of diagnostic and therapeutic findings Brain Sci Specchio N Pietrafusa N Calabrese C 7681020203311394210.3390/brainsci 10110768 PMC 7690674 · doi ↗ · pubmed ↗
- 5Oculomasticatory rhythmic movements, insomnia and stroke-like episodes in a patient with POLG mutation BMJ Case Rep Ramesh R Amanmahanya C Krishnamoorthy V Krishnan V Palani S Narasimhan Ranganathan L 017202410.1136/bcr-2023-259426 PMC 1114640638684350 · doi ↗ · pubmed ↗
- 6Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases J Inherit Metab Dis Hikmat O Naess K Engvall M 7267364320203239192910.1002/jimd.12211 · doi ↗ · pubmed ↗
- 7N Ational Library of Medicine: POLG DNA polymerase gamma, catalytic subunit [Homo sapiens (human)] 2 2025 2025 https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=Details Search&Term=5428
- 8DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations Biochim Biophys Acta Chan SS Copeland WC 312319178720091901030010.1016/j.bbabio.2008.10.007PMC 2742478 · doi ↗ · pubmed ↗