Non-malignant features of cancer predisposition syndromes manifesting in childhood and adolescence: a guide for the general pediatrician
Michaela Kuhlen, Andreas B. Weins, Nicole Stadler, Daniela Angelova-Toshkina, Michael C. Frühwald

TL;DR
This paper helps pediatricians identify non-cancer symptoms of genetic disorders that increase cancer risk in children and teens.
Contribution
The paper provides a comprehensive overview of non-malignant features of cancer predisposition syndromes for early diagnosis in pediatric patients.
Findings
Non-malignant dermatological features like café-au-lait spots and facial angiofibromas are early indicators of cancer predisposition syndromes.
Neurological and developmental anomalies, such as cerebellar ataxia and intellectual disabilities, are significant non-malignant features.
Growth and metabolic anomalies, including overgrowth and growth hormone deficiency, are notable in specific cancer predisposition syndromes.
Abstract
Cancer predisposition syndromes are genetic disorders that significantly raise the risk of developing malignancies. Although the malignant manifestations of cancer predisposition syndromes are well-studied, recognizing their non-malignant features is crucial for early diagnosis, especially in children and adolescents. A comprehensive literature search was conducted using the PubMed database, focusing on non-malignant manifestations of cancer predisposition syndromes in children and adolescents. Key sources included the Clinical Cancer Research pediatric oncology series and ORPHANET. Studies that described clinical signs and symptoms affecting specific organ systems were included. Non-malignant dermatological features often serve as early indicators of cancer predisposition syndromes, including café-au-lait spots in Neurofibromatosis Type 1 and facial angiofibromas in Tuberous…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
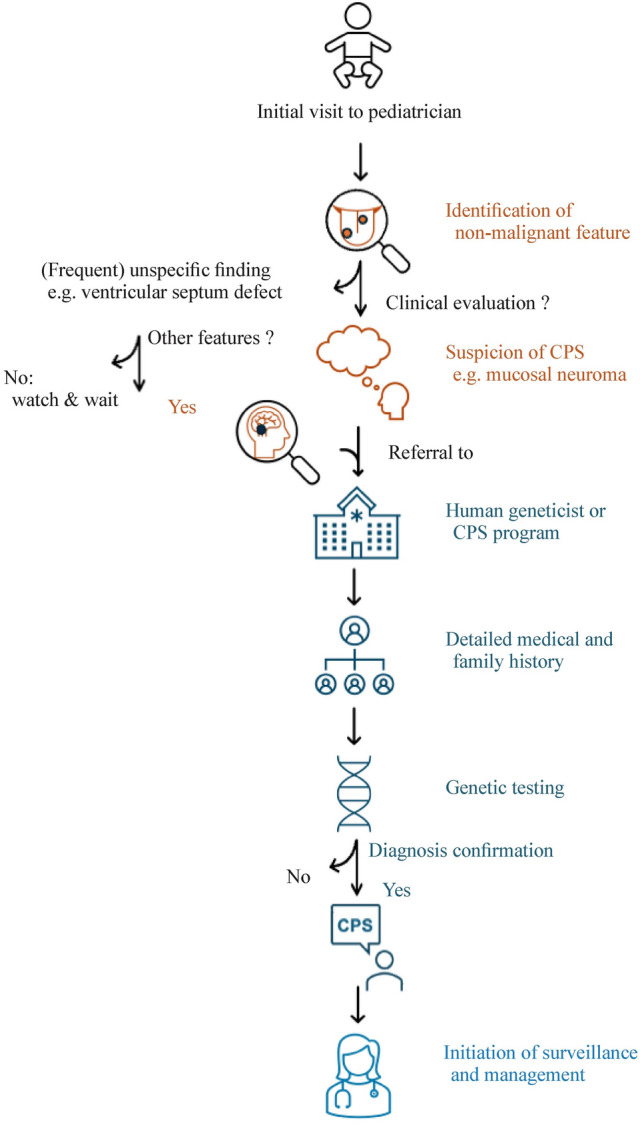 Figure 1
Figure 1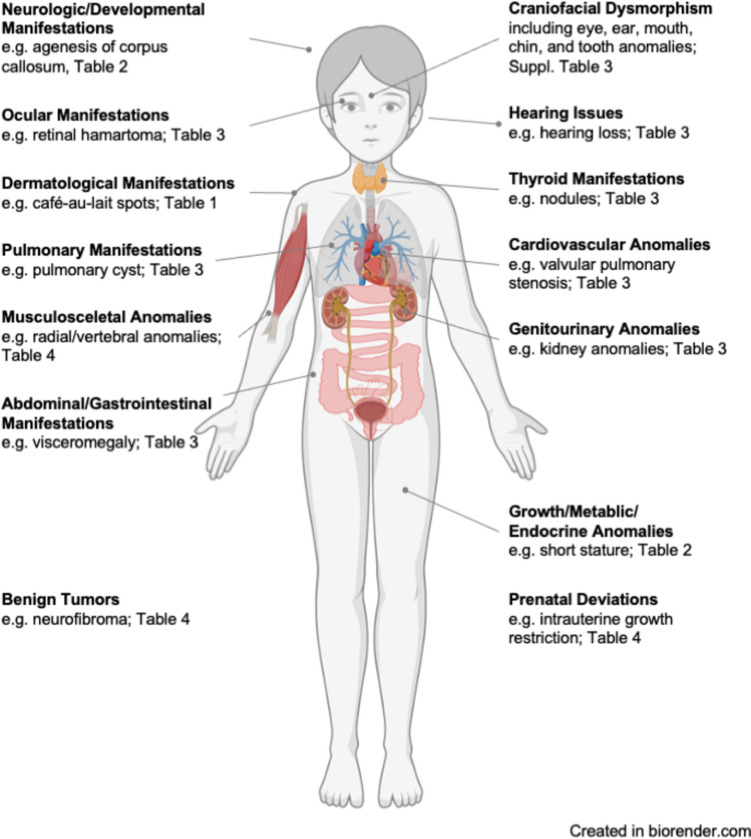 Figure 2
Figure 2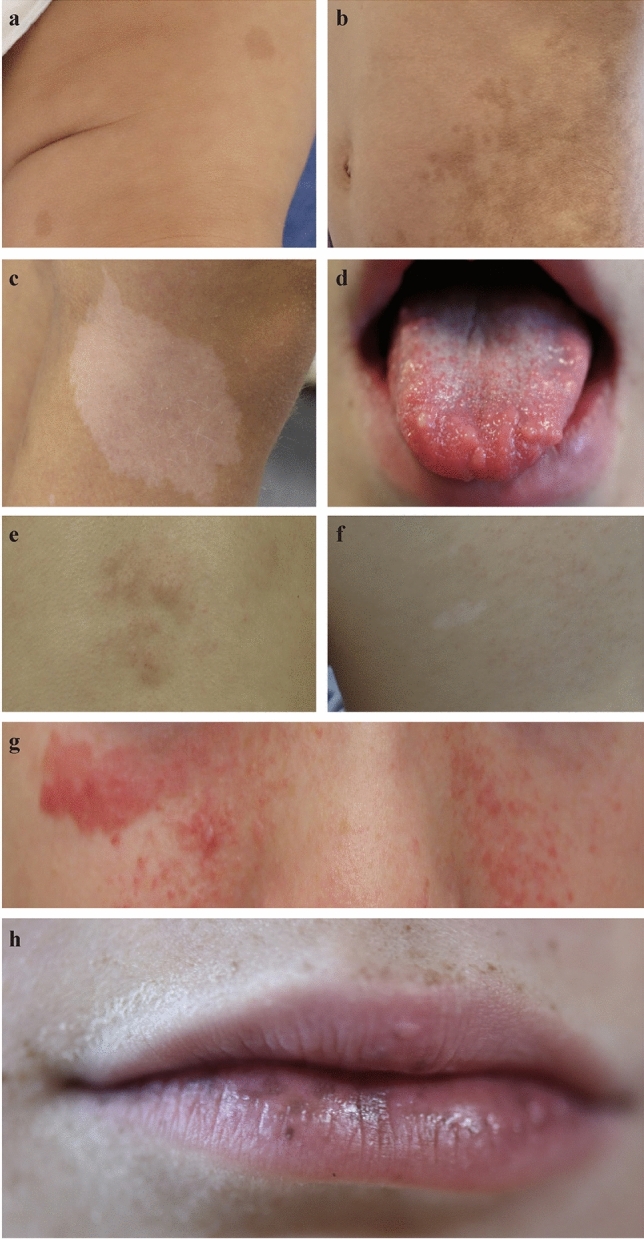 Figure 3
Figure 3 Figure 4
Figure 4- —http://dx.doi.org/10.13039/501100005972Deutsche Krebshilfe
- —http://dx.doi.org/10.13039/501100001659Deutsche Forschungsgemeinschaft
- —http://dx.doi.org/10.13039/100013081Augsburg University
- —Universität Augsburg (3144)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTuberous Sclerosis Complex Research · Tumors and Oncological Cases · Soft tissue tumor case studies
Introduction
Cancer predisposition syndromes (CPS) are a heterogeneous group of genetic disorders that significantly increase the lifetime risk of developing various malignancies [1–3]. These syndromes, often inherited in an autosomal dominant or recessive pattern, are characterized by germline variants among others in genes responsible for maintaining genomic stability, cell cycle control, DNA repair, and apoptosis [1, 4–6]. While malignant manifestations of CPS are well-documented and remain a primary focus of oncology research and clinical management [7–13], non-malignant signs and symptoms are crucial for early recognition and diagnosis [14–17].
Non-malignant manifestations encompass a wide range of clinical presentations affecting multiple organ systems, often preceding the development of malignancies or causing disabilities [14, 15, 17–19]. Pediatricians and other healthcare providers must maintain a high index of suspicion for CPS when encountering specific phenotypic anomalies, developmental delays, and other systemic manifestations in children and adolescents. Early identification of CPS through recognition of these non-malignant features facilitates timely genetic counseling, initiation of surveillance programs, and implementation of preventive measures, ultimately improving patient well-being and outcomes [1, 20].
In 2016, a workshop sponsored by the American Association for Cancer Research was held to develop consensus recommendations for cancer surveillance in children and adolescents with CPS. Experts including (co)directors of cancer predisposition programs (pediatric oncologists or medical geneticists), genetic counselors, radiologists, directors of adult cancer predisposition programs, and one pediatric endocrinologist were present. The resulting Clinical Cancer Research (CCR) pediatric oncology series provides a comprehensive overview and recommendations for surveillance of the 50 most common CPS, each carrying a 5% or greater cancer risk within the first 20 years of life [1, 20–33]. These recommendations were recently updated [34–36]. Building upon this fundamental work, the present review aims to provide an organ-specific overview of non-malignant signs and symptoms associated with CPS, offering a practical guide for daily practice (Fig. 1). This detailed exploration includes skin anomalies, neurological and developmental symptoms, growth and metabolic disorders, craniofacial dysmorphisms, ocular signs, head, neck, and thyroid anomalies, abdominal and gastrointestinal manifestations, musculoskeletal anomalies, pulmonary manifestations, cardiovascular signs, genitourinary issues, tumor development in a non-cancerous context, and prenatal deviations.Fig. 1. Flowchart of cancer predisposition syndrome (CPS) diagnosis process based on non-malignant features.This flowchart illustrates the step-by-step process for diagnosing CPS in pediatric patients, highlighting the role of recognizing non-malignant features, conducting genetic testing, and reaching a final diagnosis
By augmenting awareness of these diverse clinical presentations, this review seeks to guide pediatricians and other healthcare providers in the early detection and multidisciplinary management of children and adolescents at risk for CPS.
Methods
Literature search
We conducted a comprehensive literature search to identify relevant publications on CPS and their respective non-malignant manifestations in children and adolescents. The primary sources of data were the CCR pediatric oncology series published in 2017, covering the 50 most common CPS with ≥ 5% cancer risk (Supplemental Table 1), as well as ORPHANET and GeneReviews ® [Internet] for information on non-malignant features of these CPS. Additionally, the PubMed database was searched using combinations of the following keywords: "cancer predisposition syndromes", "non-malignant symptoms", "non-neoplastic symptoms", "phenotypic manifestations", and specific syndrome names [e.g., neurofibromatosis type 1 (NF1), Beckwith–Wiedemann syndrome (BWS)]. CPS without non-malignant manifestations, e.g. Li-Fraumeni syndrome, were excluded from this analysis.Table 1. Dermatological manifestations and associated cancer predisposition syndromesDermatological manifestationsCancer predisposition syndromeAcrochordonsBirt-Hogg-Dubé syndromeAdenomas, sebaceousMuir-Torre syndromeAdipose tissue, subcutaneous sparseBloom syndromeAlopeciaDyskeratosis congenitaAngiofibromas, facialTuberous sclerosis complexAnkle ulcerationWerner syndromeAtrophy, (sub)cutaneousRothmund-Thomson syndrome, Werner syndromeCafé-au-lait maculesBloom syndrome, cardiofaciocutaneous syndrome, constitutional mismatch repair deficiency, Fanconi anemia, NF1, Nijmegen-Breakage syndromeEczematous lesionsCardiofaciocutaneous syndrome, Shwachman-Diamond syndromeEpitheliomas, sebaceousMuir-Torre syndromeErythema (cheeks, extremities, buttocks)Rothmund-Thomson syndromeErythema, telangiectaticBloom syndromeFibrofolliculomasBirt-Hogg-Dubé syndromeFibromas, ungualTuberous sclerosis complexFrecklingConstitutional mismatch repair deficiency, NF1, xeroderma pigmentosumGranulomas, cutaneousAtaxia-TelangiectasiaHair, curlyCardiofaciocutaneous syndrome, Costello syndrome, Noonan syndrome,Hamartomas, mucocutaneousPTEN hamartoma tumor syndromeHemangiomaCardiofaciocutaneous syndromeHyperkeratosisNoonan syndrome, Rothmund-Thomson syndrome, Werner syndromeHyperpigmentationPeutz-Jeghers syndrome, Rothmund-Thomson syndromeHypertrichosisSchinzel-Giedion syndromeHypopigmentationRothmund-Thomson syndromeIchthyosisCardiofaciocutaneous syndrome, Shwachman-Diamond syndromeKeloid formationRubinstein-Taybi syndromeKeratoacanthomasMuir-Torre syndromeKeratodermaCardiofaciocutaneous syndromeLeiomyomatosis, cutaenousHereditary leiomyomatosis and renal cell cancerLentiginosis, penis/vulvaBannayan-Riley-Ruvalcaba syndromeLeukoplakia, oralDyskeratosis congenitaLichen amyloidosis, cutaneousMEN2ALipomatosis, subcutaneousBannayan-Riley-Ruvalcaba syndromeLymphedemaCardiofaciocutaneous syndrome, Noonan syndromeMacules, dark blue to brown (mouth, eyes, nares, perianal, mucosal)Peutz-Jeghers syndromeMacules, hypomelanoticTuberous sclerosis complexNail, dystrophyRothmund-Thomson syndromeNails, dysplasticDyskeratosis congenitaNail, hypoplasiaSimpson-Golabi-Behmel syndromeNails, hypoplastic/hyperconvexSchinzel-Giedion syndromeNeurofibromasConstitutional mismatch repair deficiency, NF1Neuromas, mucosal (lips, tongue)MEN2BNevus flammeusBeckwith-wiedemann syndrome, bohring-opitz syndrome, mulibrey nanismNeviCardiofaciocutaneous syndromeNevi, pigmentedNijmegen-Breakage syndromeNipples, supernumerarySimpson-Golabi-Behmel syndromeNodular tumors, subcutaneousNF2Palmar creases, singleSchinzel-Giedion syndromePapillomataCostello syndromePigmentation, reticularDyskeratosis congenitaPits, palmar/plantarGorlin syndromePlaque-like lesionsNF2Plaques, fibrousTuberous sclerosis complexSkin, dryNoonan syndrome, xeroderma pigmentosumSkin, hyperkeratotic/hyperelasticCardiofaciocutaneous syndromeSkin, loose/softCostello syndrome, Weaver syndromeSkin lesions, “confetti”Tuberous sclerosis complexSkin lesion, hyperpigmentedXeroderma pigmentosumSkin lesions, hypopigmentedBloom syndrome, xeroderma pigmentosumSkin, tightWerner syndromeTelangiectasiasAtaxia-telangiectasia, Rothmund-Thomson syndromeTrichodiscomasBirt-Hogg-Dubé syndromeVascular malformationsBannayan-Riley-Ruvalcaba syndrome, Beckwith-Wiedemann syndromeVitiligo spotsNijmegen-Breakage syndromeNF1 neurofibromatosis type 1, MEN2A Multiple Endocrine Neoplasia Type 2A, NF2 neurofibromatosis type 2
Inclusion and exclusion criteria
Studies were included whenever they met the following criteria:
- Published in English.
- Focused on non-malignant manifestations of CPS in childhood and adolescence.
- Provided detailed descriptions of clinical signs and symptoms affecting specific organ systems.
Studies were excluded if they:
- Focused solely on adult populations.
- Did not differentiate between malignant and non-malignant symptoms.
- Focused on CPS without non-malignant manifestations, e.g. Li-Fraumeni syndrome.
Data extraction and synthesis
Extracted data included syndrome names, affected organ systems, specific non-malignant signs and symptoms, frequency of signs and symptoms, and age at first occurrence, if applicable (Supplemental Table 2). No further exploration of hematological manifestations was performed. A structured approach was used to categorize the findings by organ system (Fig. 2), ensuring a comprehensive overview relevant to daily clinical practice.Table 2. Neurological, developmental, growth, metabolic, and endocrinological manifestations and associated cancer predisposition syndromesManifestationsCancer predisposition syndromeNeurological/developmental Apraxia, oculomotoricAtaxia-telangiectasia Arachnoid cystNijmegen-breakage syndrome Arnold-Chiari malformationCostello syndrome AtaxiaAtaxia-telangiectasia, xeroderma pigmentosum Attention deficit/hyperactivity disorderNoonan syndrome, tuberous sclerosis complex Autism spectrum disorderBannayan-Riley-Ruvalcaba syndrome, tuberous sclerosis complex Balance disordersAtaxia-telangiectasia Behavioral disordersSotos syndrome, WAGR syndrome ChoreoathetosisAtaxia-telangiectasia Choroid plexus hemangiomaPerlman syndrome Cognitive deficits/impairmentsNF1, xeroderma pigmentosum Coordination, poorWeaver syndrome Corpus callosum, agenesisPerlman syndrome Corpus callosum defectBohring-Opitz syndrome Cortical dysplasiaTuberous sclerosis complex Developmental delayAlagille syndrome, Bannayan-Riley-Ruvalcaba syndrome, cardiofaciocutaneous syndrome, CBL syndrome, Costello syndrome, dyskeratosis congenita, Schinzel-Giedion syndrome, Sotos syndrome DroolingAtaxia-telangiectasia DyspraxiaNoonan syndrome Epilepsy/seizuresBohring-Opitz syndrome, cardiofaciocutaneous syndrome, Costello syndrome, NF1, Schinzel-Giedion syndrome, Sotos syndrome, tuberous sclerosis complex, xeroderma pigmentosum HydrocephalusCostello syndrome, NF1, Nijmegen-Breakage syndrome Hypertonia/SpasticityWeaver syndrome, xeroderma pigmentosum HypotoniaSchinzel-Giedion syndrome, Sotos syndrome, Weaver syndrome Intellectual declineNijmegen-Breakage syndrome Intellectual disabilityCostello syndrome, Noonan syndrome, Shwachman-Diamond syndrome, Simpson-Golabi-Behmel syndrome, Seckel syndrome, Sotos syndrome, tuberous sclerosis complex, WAGR syndrome, Weaver syndrome Learning difficultiesCardiofaciocutaneous syndrome, NF1, Noonan syndrome Motor development, delayedNoonan syndrome, Simpson-Golabi-Behmel syndrome Neuropsychological deficitsTuberous sclerosis complex Psychiatric disordersTuberous sclerosis complex Psychomotor retardationShwachman-Diamond syndrome SchizencephalyNijmegen-Breakage syndrome Slurred speechAtaxia-telangiectasia Speech, delayedNoonan syndrome, Simpson-Golabi-Behmel syndrome Subependymal nodulesTuberous sclerosis complex SyringomyeliaCostello syndrome Tethered spinal cordCostello syndromeGrowth, metabolic and endocrinological ACTH, excessive productionMEN2A Bone age, advancedSotos syndrome Bone age, delayedShwachman-Diamond syndrome EndocrinopathyFanconi anemia Failure to thriveCostello syndrome, Noonan syndrome, Shwachman-Diamond syndrome Glucose intolerance/insulin resistanceAtaxia-telangiectasia, Mulibrey nanism Growth delay/deficiency/retardationAlagille syndrome, ataxia-telangiectasia, Bloom syndrome, juvenile polyposis syndrome, Nijmegen-Breakage syndrome, Noonan syndrome, Shwachman-Diamond syndrome Growth, excessiveSotos syndrome Growth failureCardiofaciocutaneous syndrome Growth hormone deficiencyCardiofaciocutaneous syndrome HemihyperplasiaBeckwith-Wiedemann syndrome HyperparathyroidismMEN1, MEN2A HypoglycemiaBeckwith-Wiedemann syndrome HypogonadismMulibrey nanism HyperinsulinismPerlman syndrome Ovarian insufficiencyNijmegen-Breakage syndrome Parathyroid hyperplasia/hypercalcemiaHyperparathyroid-jaw tumor syndrome Puberty, delayed/disorderedCostello syndrome, Frasier syndrome, Noonan syndrome Short statureCostello syndrome, Diamond-Blackfan anemia, dyskeratosis congenita, Fanconi anemia, NF1, Noonan syndrome, Shwachman-Diamond syndrome, Werner syndrome Tall statureWeaver SyndromeNF1 neurofibromatosis type 1, WAGR Wilms tumor, aniridia, genitorurinary abnormalities, and range of developmental delays syndrome, MEN2A Multiple Endocrine Neoplasia Type 2A, ATCTH Adrenocorticotropic hormoneFig. 2Organ-specific manifestations. Infographic showing which organ systems are affected by different cancer predisposition syndromes
Results
Dermatological manifestations
Non-malignant dermatological features (Table 1) are often among the earliest and most apparent indicators of CPS. Café-au-lait spots, frequently associated with NF1 [37–39] and constitutional mismatch repair deficiency (CMMRD) [15, 40, 41], are light brown skin lesions that vary in size and number (Fig. 3a, b). Patients with NF1 typically have six or more spots, which are crucial for diagnosis [37, 39]. These spots often appear in early childhood and may be accompanied by axillary or inguinal freckling. Facial angiofibromas, common to tuberous sclerosis complex (TSC) [42–45], present as red or flesh-colored papules primarily on the nose and cheeks (Fig. 3e). Other notable dermatological features include hyperpigmentation and telangiectasias in Bloom syndrome (BS) [46, 47] due to increased sun sensitivity, sebaceous adenomas in Muir–Torre syndrome (MTS) [48, 49], palmar or plantar pits in Gorlin syndrome (GS) [50–53], and mucocutaneous perioral pigmentation in Peutz–Jeghers syndrome (PJS) [54–56] (Fig. 3f). Conditions such as xeroderma pigmentosum (XP) [57, 58] and Rothmund–Thomson syndrome [59, 60] are characterized by severe photosensitivity, poikiloderma, and cutaneous atrophy, necessitating strict photoprotection to prevent skin damage and malignancy. Additionally, Birt–Hogg–Dubé syndrome (BHDS) [61] often presents with fibrofolliculomas, trichodiscomas, and acrochordons. Rubinstein–Taybi syndrome (RTS) [62] features keloid formation, while Cowden syndrome, part of PTEN hamartoma tumor syndrome (PHTS) [54, 63, 64], features pigmentation anomalies such as trichilemmomas and papillomatous papules.Fig. 3. Illustrative photos of key dermatological signs: a typical café-au-lait spots; b atypical café-au-lait spots; c hypomelanotic macule; d mucosal neuromas; e Shagreen patches; f white spots; g facial angiofibromas; h (mucocutaneous) perioral pigmentation
Neurological and developmental features
Neurological and developmental anomalies (Table 2) are significant indicators of CPS. Cerebellar ataxia, a hallmark of ataxia-telangiectasia (A-T) [65–67], manifests as a progressive loss of coordination and balance. Patients exhibit oculomotor apraxia, making eye movements difficult. Intellectual and learning disabilities are prevalent in NF1 [37–39, 68], TSC [42–45], and PHTS [54, 63, 64], posing substantial challenges during cognitive development. Seizures and autism spectrum disorder are primarily associated with TSC [42–45], with seizures often being refractory and difficult to manage. Macrocephaly is a common feature in both NF1 [37–39, 68] and PHTS [54, 63, 64], and in the context of CPS, it often coexists with other dysmorphic features or neurological anomalies. Furthermore, developmental delay, hypotonia, and speech difficulties are common to several syndromes, such as Costello syndrome (CS) [69, 70] and cardiofaciocutaneous (CFC) syndrome [71]. Patients with BS [46, 47] may demonstrate normal intelligence but suffer from speech and drooling issues that can be misinterpreted as intellectual deficiency.
Growth and metabolic anomalies
CPS can profoundly affect growth and metabolism (Table 2). BWS [72, 73] is characterized by both prenatal and postnatal overgrowth, as well as associated features such as macroglossia, which can lead to difficulties in feeding and speech. Hyperinsulinism, leading to hypoglycemia, is a common metabolic issue in BWS [72, 73]. BS [46, 47] often presents with failure to thrive due to growth difficulties, proportionate growth deficiency of prenatal onset, and continued growth deficiency throughout life. Growth hormone deficiency, often seen in NF1 [37–39] and Fanconi anemia (FA) [67, 74, 75], results in short stature and other related complications. Additionally, conditions such as Simpson–Golabi–Behmel syndrome [76–78] feature prenatal and postnatal overgrowth, macrocephaly, and macroglossia. Patients with CS [69, 70] exhibit severe postnatal feeding difficulties, failure to thrive, and short stature.
Craniofacial dysmorphism
Facial anomalies (Supplemental Table 3) are common non-malignant indicators of many CPS and can serve as important diagnostic clues. Proteus syndrome (PS) [54, 79, 80] can result in asymmetric overgrowth of facial features, contributing to a distinctive facial appearance. In multiple endocrine neoplasia type 2B (MEN2B) [17, 81, 82], mucosal neuromas (Fig. 3d) cause bumpy, often enlarged lips and tongue.Table 3. Organ-specific manifestations including ocular manifestations, hearing issues, dental issues, thyroid, pulmonary, cardiovascular, abdominal/gastrointestinal, and genitourinary manifestations and associated cancer predisposition syndromesManifestationsCancer predisposition syndromeOcular manifestations AlacrimaMEN2B AniridiaWAGR syndrome Axenfeld anomalyAlagille syndrome BlepharitisDyskeratosis congenita CataractGorlin syndrome, NF2, WAGR syndrome, Werner syndrome ColobomaGorlin syndrome Corneal nerve, prominentMEN2B Corneal opacification/vascularizationWAGR syndrome Embryotoxon, posteriorAlagille syndrome EpiphoraDyskeratosis congenita Glaucoma (congenital)Rubinstein-Taybi syndrome, WAGR syndrome Iris hamartomasNF1 KeratitisXeroderma pigmentosum MicrophthalmusFanconi anemia, Gorlin syndrome MyopiaBohring-Opitz syndrome Nasolacrimal duct obstructionRubinstein-Taybi syndrome NystagmusCardiofaciocutaneous syndrome Optic nerve hypoplasiaWAGR syndrome PhotophobiaXeroderma pigmentosum Retinal hamartomaTuberous sclerosis complex Retinal hemangioblastomaVon-Hippel-Lindau syndrome Retinal/optic nerve anomaliesBohring-Opitz syndrome, cardiofaciocutaneous syndrome Retinal mid-peripheral region, yellowish dotsMulibrey nanism Retinal pigment epithelium, hypertrophyFamilial adenomatous polyposis Retinopathy, pigmentaryAlagille syndrome Papillary and optic disc anomaliesAlagille syndrome StrabismusCardiofaciocutaneous syndrome, Noonan syndrome Telangiectasias, retinalAtaxia-telangiectasia Visual impairmentSchinzel-Giedion syndromeHearing issues DeafnessNF2 Endolymphatic sac tumorVon-Hippel-Lindau syndrome Hearing lossFanconi anemia, NF2, Noonan syndrome, Sotos syndrome, xeroderma pigmentosum Hearing impairmentSchinzel-Giedion syndromeDental issues Dental anomaliesRothmund-Thomson syndrome, Shwachman-Diamond syndrome Dental crowdingNoonan syndrome Dental enamel pittingTuberous sclerosis complex Dental eruption, prematureSotos syndrome Dental malocclusionSimpson-Golabi-Behmel syndrome Dentigerous cystsFamilial adenomatous polyposis Jaw fibromas, ossifyingHyperparathyroid-jaw tumor syndrome Keratocysts, mandibular odontogenicGorlin syndrome Periodontal diseaseDyskeratosis congenita TaurodontismDyskeratosis congenita Teeth/root ratio, decreasedDyskeratosis congenita Teeth, supernumeraryFamilial adenomatous polyposis Teeth, uneruptedFamilial adenomatous polyposisThyroid manifestations Hashimoto thyroiditisBannayan-Riley-Ruvalcaba syndrome, PTEN hamartoma tumor syndrome HypothyroidismAlagille syndrome Thyroid dysfunctionNoonan syndrome Thyroid dysplasiaDICER1 syndrome Thyroid nodulesDICER1 syndrome, PTEN hamartoma tumor syndromePulmonary manifestations Airway infections, recurrentAtaxia-telangiectasia, Nijmegen-Breakage syndrome, Rubinstein-Taybi syndrome BronchiectasisAtaxia-telangiectasia ChylothoraxTuberous sclerosis complex Cysts, (sub)pleuralBirt-Hogg-Dubé syndrome Diaphragmatic herniaPerlman syndrome, Simpson-Golabi-Behmel syndrome LymphangioleiomyomatosisTuberous sclerosis complex Pneumocyte hyperplasia, multifocal micronodularTuberous sclerosis complex PneumothoraxBirt-Hogg-Dubé syndrome, tuberous sclerosis complex Pulmonary arteriovenous malformationsDyskeratosis congenita Pulmonary cystDICER1 syndrome, tuberous sclerosis complex Pulmonary fibrosisDyskeratosis congenita Sleep apnea, ObstructiveBohring-Opitz syndromeCardiovascular anomalies Aortic arch, interruptedPerlman syndrome ArrhythmiaCostello syndrome, Simpson-Golabi-Behmel syndrome Cardiac malformationsBeckwith-Wiedemann syndrome, Simpson-Golabi-Behmel syndrome, Sotos syndrome Cardiomyopathy, hypertrophicCardiofaciocutaneous syndrome, Coronary arteries, dilatationNoonan syndrome Dextroposition, heartPerlman syndrome Ductus arteriosus, patentAlagille syndrome, Rubinstein-Taybi syndrome, Schinzel-Giedion syndrome HypertensionNF1 Moyamoya diseaseNF1, Noonan syndrome Perimyocardial heart disease, restrictiveMulibrey nanism Pulmonary artery stenosis or atresiaAlagille syndrome Septal defects, atrial and/or ventricularAlagille syndrome, Bohring-Opitz syndrome, Noonan syndrome, Rubinstein-Taybi syndrome, Schinzel-Giedion syndrome Tetralogy of FallotAlagille syndrome Valvular dysplasiaSchinzel-Giedion syndrome Valvular pulmonary stenosisCardiofaciocutaneous syndrome, Costello syndrome, Noonan syndrome Vascular malformationPTEN hamartoma tumor syndrome Ventricles, hypoplasticSchinzel-Giedion syndromeAbdominal/gastrointestinal manifestations Anorectal malformationsFanconi anemia Cholestasis, chronicAlagille syndrome Constipation, chronicCardiofaciocutaneous syndrome, MEN2B, Rubinstein-Taybi syndrome DiarrheaMEN2A, MEN2B Diastasis rectiBeckwith-Wiedemann syndrome Duodenal atresiaFanconi anemia Esophageal atresiaFanconi anemia Esophageal stenosisDyskeratosis congenita Ganglioneuromatosis, generalizedMEN2B Hepatic fibrosisPerlman syndrome HepatomegalyMulibrey nanism Hernia, umbilicalBeckwith-Wiedemann syndrome, Simpson-Golabi-Behmel syndrome, Weaver syndrome Hirschsprung diseaseMEN2A Ileal atresia, distalPerlman syndrome Liver diseaseDyskeratosis congenita Liver, fattyMulibrey nanism MegacolonMEN2B Pancreatic cystsVon-Hippel-Lindau syndrome Pancreatic insufficiency, exocrineShwachman-Diamond syndrome SteatohepatitisAtaxia-telangiectasia Telangiectasias, gastrointestinalDyskeratosis congenita VisceromegalyBeckwith-Wiedemann syndrome, Perlman syndrome, Simpson-Golabi-Behmel syndromeGenitourinary anomalies Anus, anteriorly displacedSchinzel-Giedion syndrome Azo-/oligospermiaBloom syndrome CryptorchidismNijmegen-Breakage syndrome, Noonan syndrome, Perlman syndrome, Schinzel-Giedion syndrome, Simpson-Golabi-Behmel syndrome Epididymal cysts/cystadenomasVon-Hippel-Lindau syndrome Genitalia, ambiguous externalDenys-Drash syndrome Gonadal dysgenesisFrasier syndrome HydronephrosisSchinzel-Giedion syndrome Hypoplastic labia majora/minoraSchinzel-Giedion syndrome Hypoplastic uterusSchinzel-Giedion syndrome HypospadiasNijmegen-Breakage syndrome, Schinzel-Giedion syndrome, Simpson-Golabi-Behmel syndrome Impaired fertility/infertilityAtaxia-telangiectasia, Fanconi anemia, Frasier syndrome, Mulibrey nanism, Noonan syndrome Labial sulcus, deepSchinzel-Giedion syndrome Kidney, ectopic/dystopicNijmegen-Breakage syndrome Kidney, HorseshoeNijmegen-Breakage syndrome Kidneys, small/dysplasticAlagille syndrome, Simpson-Golabi-Behmel syndrome Kidney-related anomaliesBeckwith-Wiedemann syndrome, Denys-Drash syndrome MicropenisSchinzel-Giedion syndrome Nephroma, cysticDICER1 syndrome NephropathyFrasier syndrome Nephrotic syndromeDenys-Drash syndrome, Frasier syndrome Ovary hypoplasiaNijmegen-Breakage syndrome Renal cystsHyperparathyroid-jaw tumor syndrome, Von-Hippel-Lindau syndrome Renal disease, end-stageFrasier syndrome, Hyperparathyroid-jaw tumor syndrome, WAGR syndrome Testicular development, disorderDenys-Drash syndrome Urethral stenosisDyskeratosis congenita Urogenital anomaliesDiamond-Blackfan anemia, Sotos syndrome, WAGR Syndrome UrolithiasisHyperparathyroid-jaw tumor syndromeNF1 neurofibromatosis type 1, NF2 neurofibromatosis type 2, MEN2A Multiple Endocrine Neoplasia Type 2A, MEN2B Multiple Endocrine Neoplasia Type 2B
A broad or prominent forehead is observed in several CPS. For instance, patients with Noonan syndrome (NS) [83, 84] typically present with a broad forehead, while those with CFC syndrome [71] exhibit a large forehead. Bohring–Opitz syndrome (BOS) [85] also includes a prominent forehead with glabellar nevus flammeus. RTS [62] is characterized by highly arched eyebrows and long eyelashes. Patients with NS [83, 84] may present with hypertelorism and ptosis, as well as downslanting palpebral fissures. In CS [69, 70], ptosis and full cheeks are common, while BOS [85] features synophrys, proptosis, and hypertelorism. A convex nasal ridge is typical in RTS [62], whereas patients with CFC syndrome [71] often present with a short nose with a depressed nasal bridge. BOS [85] can feature a depressed, wide nasal bridge and anteverted nares. NS [83, 84] patients frequently have a short nose with a depressed nasal bridge as well. Patients with CS [69, 70] often have a large mouth with prominent lips and thickened ear helices. Ear anomalies are also notable in several CPS; low-set, posteriorly rotated ears are common in NS [83, 84], while patients with CFC syndrome [71] may exhibit low-set ears as well.
Ocular manifestations
Non-malignant ocular symptoms (Table 3) include retinal hamartomas in TSC [42–45] and Lisch nodules, which are pigmented iris hamartomas, in NF1 [37–39]. Patients with Wilms tumor-aniridia-genitourinary anomalies-intellectual disability (WAGR) syndrome [86, 87] frequently exhibit aniridia, which can be associated with other eye anomalies such as cataract, glaucoma, and optic nerve hypoplasia. Additionally, conditions like BOS [85] can present with retinal and optic nerve anomalies, high myopia, and other vision issues.
Hearing issues
Hearing loss (Table 3), both sensorineural and conductive, is associated with neurofibromatosis type 2 (NF2) [88–90]. Patients with NF2 often develop bilateral vestibular schwannomas, leading to progressive hearing loss.
Thyroid manifestations
Thyroid anomalies (Table 3) are prevalent in several CPS. For example, PHTS [54, 63, 64] often presents with thyroid nodules and a predisposition to thyroid cancer. DICER1 syndrome [64, 91, 92] is associated with multinodular goiter and differentiated thyroid cancer.
Pulmonary manifestations
Pulmonary cysts leading to spontaneous pneumothorax are characteristic of BHDS [61]. In DICER1 syndrome [64, 91, 92], cystic lesions in terms of pleuropulmonary blastoma (PPB) type I may precede the more aggressive malignant types II and III. Chronic respiratory infections due to immunodeficiency are common in A-T [65–67], and pulmonary fibrosis is a significant concern in dyskeratosis congenita [93, 94]. Patients with TSC [42–45] may develop lymphangioleiomyomatosis and multifocal micronodular pneumocyte hyperplasia.
Cardiovascular anomalies
Congenital heart defects such as pulmonary valve stenosis and hypertrophic cardiomyopathy are associated with NS [83, 84] and CS [69, 70]. Patients with CFC syndrome [71] often present with valvular pulmonary stenosis and hypertrophic cardiomyopathy. Vascular anomalies, including arterial stenosis and aneurysms, are notable in NF1 [37–39, 68] and BS [46, 47], whereas cardiac fibromas are typical in GS [50–53]. Vascular malformations are also significant, with conditions like Bannayan–Riley–Ruvalcaba syndrome [54, 63, 95] presenting with hemangiomas and vascular anomalies, and PS [54, 79, 80] featuring vascular malformations, which may be capillary, venous, or lymphatic in nature.
Abdominal and gastrointestinal manifestations
Hepatomegaly and nephromegaly are frequent findings in BWS [72, 73], while splenomegaly is common in FA [67, 74, 75]. Additionally, gastrointestinal anomalies such as esophageal and duodenal atresia may occur in individuals with FA, contributing to feeding difficulties and requiring surgical interventions early in life. Cystic nephromas, which are benign kidney tumors, are often seen in DICER1 syndrome [64, 91, 92]. PJS [54–56] presents with gastrointestinal polyps, which may be detected at any site within the GI tract, most frequently in the small intestine, and may lead to complications such as intussusception.
Genitourinary anomalies
Renal cysts and tumors are prevalent in Von Hippel-Lindau (VHL) disease [96–98] and BHDS [61]. Ambiguous genitalia and disorders of sex development are indicative of WAGR syndrome [86, 87]. Denys–Drash syndrome [99] features nephropathy that progresses to end-stage renal disease, along with genital anomalies such as ambiguous genitalia.
Musculoskeletal anomalies
Musculoskeletal anomalies (Table 4) include scoliosis and bone dysplasia in NF1 [37–39], and jaw cysts along with bifid ribs in GS [50–53]. MEN2B [17, 81, 82] is characterized by marfanoid habitus, pes cavus, pectus excavatum, and joint hyperextensibility. RTS [62] features joint hypermobility and skeletal dysplasia.Table 4. Musculoskeletal anomalies, benign tumors, and prenatal deviations and associated cancer predisposition syndromesManifestationsCancer predisposition syndromeMusculoskeletal anomalies Achilles tendons, tightCostello syndrome Bones, slender long with thick cortex, narrow medullary channelMulibrey nanism BrachydactylySimpson-Golabi-Behmel syndrome Brachymelia, mesomelicSchinzel-Giedion syndrome CamptodactyliaWeaver syndrome ClinodactyliaWeaver syndrome ContracturesBohring-Opitz syndrome Elbows, flexionBohring-Opitz syndrome Elbow, radial head dislocationBohring-Opitz syndrome Extremities, hypertonicBohring-Opitz syndrome Femoral epiphysis, slipped capitalMEN2B Fibrous dysplasiaMulibrey nanism Foot deformitySchinzel-Giedion syndrome Hip dislocation/dysplasiaBohring-Opitz syndrome, Costello syndrome HypotoniaBohring-Opitz syndrome, cardiofaciocutaneous syndrome, CBL syndrome Joints, hyperextensible/hypermobileMEN2B, Rubinstein-Taybi syndrome Joint laxityCostello syndrome, Weaver syndrome Limbs, shortSchinzel-Giedion syndrome Marfanoid habitusMEN2B Metaphyseal dysplasiaShwachman-Diamond syndrome Muscular hypoplasia, abdominalPerlman syndrome MyopathyBannayan-Riley-Ruvalcaba syndrome Osteoporosis/osteopeniaCostello syndrome, dyskeratosis congenita, NF1, Shwachman-Diamond syndrome, Werner syndrome Pectus carinatumShwachman-Diamond syndrome Pectus excavatumBannayan-Riley-Ruvalcaba syndrome, MEN2B, Weaver syndrome Pectus deformityGorlin syndrome Pes cavusMEN2B PolydactylyGorlin syndrome, Simpson-Golabi-Behmel syndrome PseudoarthrosisNF1 Radial/thumb anomaliesDiamond-Blackfan anemia, Fanconi anemia Rib anomaliesGorlin syndrome Skeletal dysplasiaTuberous sclerosis complex ScoliosisCostello syndrome, Gorlin syndrome, MEN2B, NF1, Noonan syndrome, Sotos syndrome, Weaver syndrome Sella turcica, J-shapedMulibrey nanism Shoulders, internal rotationBohring-Opitz syndrome Sphenoid wingNF1 Sprengel deformityGorlin syndrome Sternal deformityNoonan syndrome SyndactylyGorlin syndrome, Simpson-Golabi-Behmel syndrome Talipes equinovarusNoonan syndrome Thoracic cage, smallMulibrey nanism Ulnar deviation of wrist, fingersBohring-Opitz syndrome, Costello syndrome Vertebral anomaliesAlagille syndrome, Gorlin syndrome, NF1, Simpson-Golabi-Behmel syndromeBenign tumors AdenomaConstitutional mismatch repair deficiency Adenoma, parathyroidHyperparathyroid-jaw tumor syndrome Angiomyolipomas, renalTuberous sclerosis complex Desmoid tumorFamilial adenomatous polyposis Fibroelastoma, heartGorlin syndrome Fibroma, intraoralTuberous sclerosis complex Fibroma, ovarianGorlin syndrome Giant cell astrocytoma, subependymalTuberous sclerosis complex HamartomaHyperparathyroid-jaw tumor syndrome, tuberous sclerosis complex HemangioblastomaVon-Hippel-Lindau syndrome Leiomyoma, cutaneous/uterineHereditary leiomyomatosis and renal cell cancer Melanoma, ocularXeroderma pigmentosum MeningeomaGorlin syndrome, NF2 Neurofibromas, plexiformNF1 Neuroendocrine tumorsMEN1, tuberous sclerosis complex, Von-Hippel-Lindau syndrome Oncocytoma, renalBirt-Hogg-Dubé syndrome Optic pathway gliomaNF1 OsteomaFamilial adenomatous polyposis Pituitary tumorsMEN1 Polyposis, gastrointestinal hamartomatousBannayan-Riley-Ruvalcaba syndrome PolypConstitutional mismatch repair deficiency, familial adenomatous polyposis, Hyperparathyroid-Jaw tumor syndrome, juvenile polyposis syndrome, PTEN hamartoma tumor syndrome Polyps, hamartomatousPeutz-Jeghers syndrome Rhabdomyomas, cardiacTuberous sclerosis complex SchwannomasNF2 Sex cord tumors with annular tubulesPeutz-Jeghers syndromePerinatal deviations Birth length, highWeaver syndrome Birth length, lowFanconi anemia Birth weight, highCostello syndrome, Weaver syndrome Birth weight, lowFanconi anemia Dwarfism, prenatal onsetSeckel syndrome Fetal adrenocortical cytomegalyBeckwith-Wiedemann syndrome Growth, excessive, intrauterineSotos syndrome Growth restriction, intrauterineBohring-Opitz syndrome, Mulibrey nanism HydropsCostello syndrome OmphaloceleBeckwith-Wiedemann syndrome PolyhydramniosCardiofaciocutaneous syndromeNF1 neurofibromatosis type 1, NF2 neurofibromatosis type 2, MEN1 Multiple Endocrine Neoplasia Type 1, MEN2B Multiple Endocrine Neoplasia Type 2B, CBL Casitas B-lineage lymphoma syndrome
Benign tumors
Benign tumors (Table 4), including lipomas, hamartomas, and adenomas, are commonly observed in MEN syndromes and PJS [54–56]. MTS [48, 49] features sebaceous adenomas and epitheliomas, while PHTS [54, 63, 64] presents with mucocutaneous hamartomas and thyroid pathology.
Prenatal deviations
Polyhydramnios and preterm birth represent obstetric complications associated with BWS [72, 73] and TSC [42–45]. BWS [72, 73] may be associated with abdominal wall defects.
Discussion
CPS present a multifaceted clinical challenge due to their broad spectrum of non-malignant and malignant manifestations. These syndromes, which include conditions such as NF1, TSC, and BWS, among others, often require a high index of suspicion and an interdisciplinary approach to care.
Complexity of non-malignant manifestations
The non-malignant features of CPS affect nearly every organ system, necessitating comprehensive and continued evaluation (Fig. 2). For instance, dermatological manifestations such as café-au-lait spots, facial angiofibromas, and sebaceous adenomas serve as critical early indicators that should prompt further genetic evaluation (Fig. 1) [16, 19, 37, 44, 45, 49, 100]. These features are not merely cosmetic concerns but are pivotal in the early diagnosis of CPS, facilitating timely intervention and management [68, 101].
Neurological and developmental symptoms, such as intellectual disabilities, learning challenges, and cerebellar ataxia, significantly impact the quality of life and developmental trajectory of affected individuals. For instance, cerebellar ataxia in A-T leads to progressive loss of coordination and balance, severely affecting daily functioning [65, 66]. Intellectual disabilities in NF1 and TSC result in substantial cognitive challenges, highlighting the need for continuous monitoring and specialized educational support [45, 68, 100].
Growth and metabolic anomalies, including prenatal and postnatal overgrowth seen in BWS and growth hormone deficiency in various CPS, require close collaboration between endocrinologists, geneticists, and nutritionists [73, 102]. Effective management of these conditions has the potential to prevent severe complications and improve overall outcomes. For instance, hyperinsulinism in BWS can lead to life-threatening hypoglycemia, highlighting the need for vigilant metabolic monitoring and management.
Importance of interdisciplinary care
The multi-system involvement characteristic of CPS underscores the necessity for interdisciplinary care [37, 96, 103, 104]. This approach ensures comprehensive management of both the non-malignant and malignant aspects of these syndromes. Pediatricians, dermatologists, neurologists, endocrinologists, cardiologists, geneticists, and other specialists must collaborate closely to provide holistic care tailored to the individual needs of each patient.
For instance, the management of NF1 requires regular assessments to monitor neurofibroma, alongside neurological evaluations to address cognitive and developmental issues [37, 39, 68, 100]. Similarly, patients with A-T benefit from coordinated care involving immunologists for immunodeficiency management and infection prophylaxis, neurologists for monitoring neurological symptoms, and oncologists for ongoing tumor surveillance [65–67].
Moreover, genetic counseling plays a crucial role in the care of families affected by CPS [20]. It helps in understanding the hereditary nature of these syndromes, provides risk assessments for family members, and informs reproductive decisions. Genetic counselors help navigate families through the complexities of genetic testing and the implications of the results.
Surveillance and preventive measures
Early identification and regular surveillance are essential in managing CPS [105–107]. These measures enable the early detection of complications and help prevent the progression of symptoms. For example, regular MRI scans are essential for detecting and monitoring brain tumors in patients with SUFU-associated GS, while ultrasound screenings are vital for identifying abdominal tumors in BWS [23, 51, 73].
Preventive measures, including strict photoprotection in conditions like BS and XP, are vital in reducing the risk of skin cancers [67]. Additionally, prophylactic interventions such as thyroidectomy in MEN2 to prevent medullary thyroid carcinoma, exemplify proactive management strategies necessary in CPS care [14, 33].
Psychosocial support
The psychosocial impact of CPS on patients and their families is significant [108]. Chronic conditions and the potential for malignancies pose substantial emotional and psychological challenges [109, 110]. Providing comprehensive psychosocial support, including counseling and access to support groups, is crucial in helping patients and their families cope with the stress and uncertainties associated with CPS [110, 111].
Transition aspects for children and adolescents with cancer predisposition syndromes
Transitioning from pediatric to adult care is a critical period for children and adolescents with CPS [112, 113]. This transition must be meticulously planned and managed to ensure continuity of care, adherence to surveillance protocols, and psychosocial support. Adolescents with CPS often face unique challenges due to the lifelong nature of their conditions, the complexity of their medical needs, and the potential for both malignant and non-malignant manifestations. Effective transition planning should start early, involving a multidisciplinary team that includes pediatricians, oncologists, and adult care providers. This ensures that all aspects of the patient’s health are considered and addressed. Education about the genetic nature of CPS, potential health risks, and the importance of continued surveillance empowers patients to understand their condition better, promoting self-management and adherence to medical recommendations. A well-structured transition program can significantly improve health outcomes and quality of life for adolescents with CPS, fostering a smooth and effective shift from pediatric to adult healthcare systems.
In conclusion, this comprehensive overview highlights the diverse range of non-malignant manifestations associated with CPS, emphasizing the critical importance of early detection and multidisciplinary management. By recognizing these features, pediatricians and other healthcare providers facilitate timely diagnosis, genetic counselling, and appropriate surveillance. This proactive approach ultimately improves medical outcomes and the well-being of affected children, adolescents, and those at risk of CPS.
Supplementary Information
Below is the link to the electronic supplementary material.Supplementary file1 (DOCX 117 kb)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Goudie C, Coltin H, Witkowski L, Mourad S, Malkin D, Foulkes WD. The Mc Gill Interactive Pediatric Onco Genetic Guidelines: an approach to identifying pediatric oncology patients most likely to benefit from a genetic evaluation. Pediatr Blood Cancer. 2017;64.10.1002/pbc.2644128097779 · doi ↗ · pubmed ↗
- 2Kamihara J, Diller LR, Foulkes WD, Michaeli O, Nakano Y, Pajtler KW, et al. Neuroblastoma Predisposition and Surveillance-An Update from the 2023 AACR Childhood Cancer Predisposition Workshop. Clin Cancer Res. 2024:OF 1-OF 7.10.1158/1078-0432.CCR-24-023738860978 · doi ↗ · pubmed ↗
- 3Hafsi W, Badri T, Rice AS. Bloom syndrome. Treasure Island (FL): Stat Pearls; 2024.28846287 · pubmed ↗
- 4Gay JT, Troxell T, Gross GP. Muir-Torre syndrome. Treasure Island (FL): Stat Pearls; 2024.30020643 · pubmed ↗
- 5Evans D. Nevoid basal cell carcinoma syndrome. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington; 2002 [Updated 2024 Feb 22].20301330 · pubmed ↗
- 6Kraemer K, Di Giovanna J, Tamura D. Xeroderma pigmentosum. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington; 2003 [Updated 2022 Mar 24].20301571 · pubmed ↗
- 7Stevens C. Rubinstein-Taybi syndrome. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington; 2002 [Updated 2023 Nov 9].20301699 · pubmed ↗
- 8Mehta P, Ebens C. Fanconi anemia. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington; 2021.20301575 · pubmed ↗