The SEP homologous gene TEMARY regulates inflorescence phenotypes in Hydrangea Macrophylla
Kenji Nashima, Tatsuya Uemachi, Kenta Shirasawa, Akifumi Shimizu, Toshiki Takeuchi, Tatsuya Obata, Sachiko Isobe, Mirai Azuma, Midori Akutsu, Yoshiko Nakazawa, Masaharu Kodama, Kiyoshi Namai, Takeshi Kurokura, Takuro Suyama

TL;DR
This study identifies the TEMARY gene as responsible for the mophead inflorescence phenotype in Hydrangea macrophylla, showing how mutations in this gene lead to changes in flower arrangement.
Contribution
The study discovers TEMARY, a SEP homologous gene, as the key regulator of inflorescence type in Hydrangea macrophylla.
Findings
Three types of loss-of-function TEMARY alleles were identified in mophead cultivars.
Mophead mutants do not express TEMARY normally, as shown by RNA-seq and qRT-PCR analyses.
The TEMARY gene locus in mophead cultivars consists only of loss-of-function alleles.
Abstract
Most Hydrangea species have inflorescences composed of two types of flowers: decorative flowers with showy sepals and plain nondecorative flowers. In wild plants of Hydrangea macrophylla, the decorative flowers are located at the periphery of the corymb, resulting in the lacecap phenotype. However, after the discovery of the mophead phenotype, in which decorative flowers are borne not only at the periphery but also on the entire inflorescence, it developed remarkably as a garden plant. In this study, we aimed to identify the gene controlling the inflorescence type and the mutations causing the mophead phenotype. Linkage analyses identified a SEPALLATA (SEP) homologous gene as a candidate gene, named TEMARY. We analyzed the genome sequences of TEMARY using several cultivars. The results revealed that the H. macrophylla cultivars had three types of loss-of-function alleles, and that the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
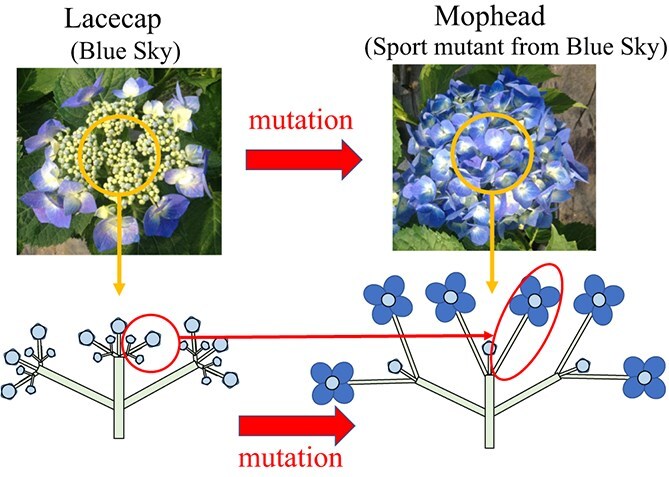 Figure 1
Figure 1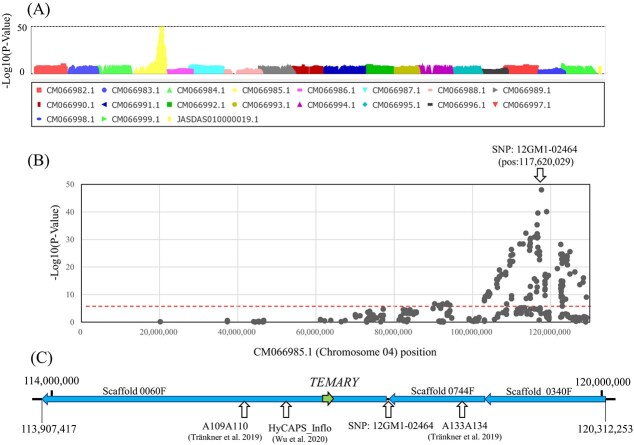 Figure 2
Figure 2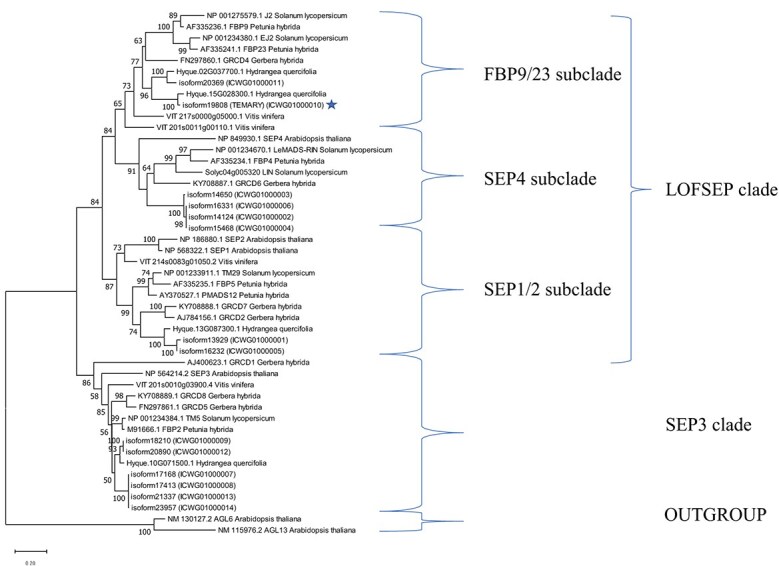 Figure 3
Figure 3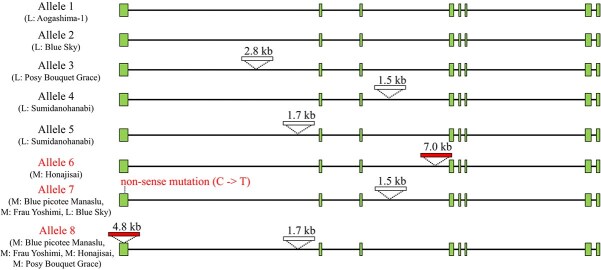 Figure 4
Figure 4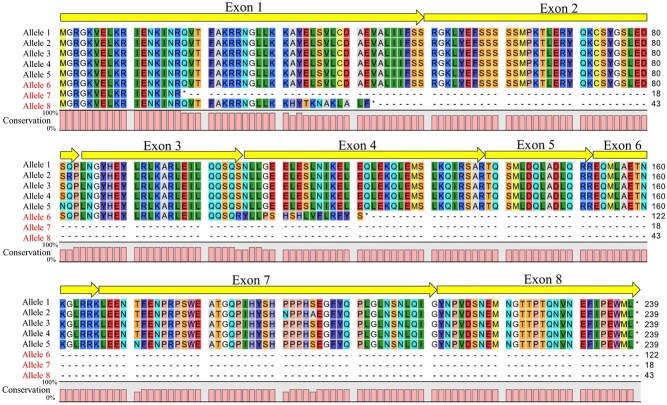 Figure 5
Figure 5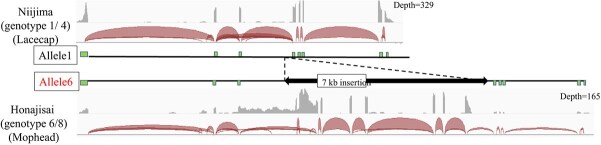 Figure 6
Figure 6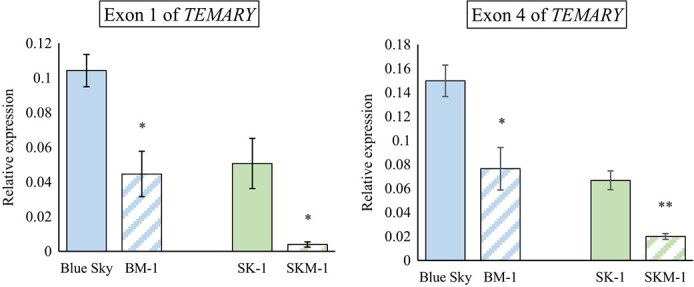 Figure 7
Figure 7| RNA-Seq expression (FPKM) | ||||||
|---|---|---|---|---|---|---|
| Gene name in Hma_r1.2_1 |
Best hit | SK-1 | SKM-1 | BlueSky | BM-1 | |
| Hma1.2p1_0060F.1_g033600 | At3g01140 | Transcription factor MYB106 | 0.72 | 0 | 0.43 | 0 |
| Hma1.2p1_0060F.1_g033470 | At3g01090 | SNF1-related protein kinase catalytic subunit alpha KIN10 | 21.95 | 6.3 | 21.7 | 6.04 |
| Hma1.2p1_0060F.1_g033330 | At3g44900 | Cation/H(+) antiporter 4 | 0.17 | 0 | 0.08 | 0 |
| Hma1.2p1_0060F.1_g032830 | At4g18372 | Small nuclear ribonucleoprotein family protein | 3.47 | 0 | 6.17 | 4.47 |
| Hma1.2p1_0060F.1_g032780 | At5g15800 | Developmental protein SEPALLATA 1 | 14.22 | 1.6 | 24.29 | 10.99 |
| Hma1.2p1_0340F.1_g130010 | At1g74070 | Peptidyl-prolyl cis-trans isomerase CYP26–2 | 3.39 | 2.38 | 0.69 | 0 |
| Hma1.2p1_0340F.1_g130020 | At1g74070 | Peptidyl-prolyl cis-trans isomerase CYP26–2 | 1.12 | 0.65 | 0.21 | 0 |
| Hma1.2p1_0340F.1_g129940 | At1g49000 | Transmembrane protein | 2.53 | 3.12 | 0.28 | 0.09 |
| Hma1.2p1_0340F.1_g129810 | AT5g38690 | Zinc-finger domain of monoamine-oxidase A repressor R1 protein | 16.94 | 7.41 | 5.58 | 1.3 |
| Hma1.2p1_0060F.1_g032930 | At1g74110 | Cytochrome P450 family protein CYP78A10 | 293.52 | 272.72 | 442.38 | 281.04 |
| FPKM | ||||
|---|---|---|---|---|
| Accession | Phenotype | Allele 2 | Allele 7 | Allele 8 |
| Blue Sky | Lacecap | 13.59 | 12.35 | - |
| BM-1 (sport mutant of BlueSky) | Mophead | 2.39 | 9.51 | - |
| SK-1 | Lacecap | 17.60 | - | 0.11 |
| SKM-1 (sport mutant of SK-1) | Mophead | 2.72 | - | 0.14 |
| Accession | Phenotype | Genotype | |
|---|---|---|---|
| Amethyst | Mophead | 7 | 8 |
| Benelux | Mophead | 7 | 8 |
| Blue Picotee Manaslu | Mophead | 7 | 8 |
| Bodensee | Mophead | 7 | 8 |
| Chibori | Mophead | 8 | 8 |
| Chikushi Ruby | Mophead | 7 | 8 |
| Chikushinomai | Mophead | 7 | 8 |
| Felix | Mophead | 6 | 8 |
| Flambeau | Mophead | 6 | 8 |
| Frau Mariko | Mophead | 7 | 8 |
| Frau Yoshiko | Mophead | 8 | 8 |
| Frau Yoshimi | Mophead | 7 | 8 |
| Green Shadow | Mophead | 8 | 8 |
| Honajisai | Mophead | 6 | 8 |
| Immaculata | Mophead | 6 | 8 |
| Junihitoe | Mophead | 6 | 8 |
| Kanuma Blue | Mophead | 8 | 8 |
| Madame Emile Mouillere | Mophead | 8 | 8 |
| Mathilde Gutges | Mophead | 8 | 8 |
| Mausseline | Mophead | 7 | 8 |
| Mis. Kumiko | Mophead | 8 | 8 |
| Paris | Mophead | 7 | 8 |
| Peach Hime | Mophead | 8 | 8 |
| Ruby Red | Mophead | 7 | 8 |
| Sensation | Mophead | 8 | 8 |
| Shinkai | Mophead | 7 | 7 |
| Uzuajisai | Mophead | 6 | 8 |
| 03JP1 | Lacecap | 3 | 7 |
| 12GM1 | Lacecap | 3 | 8 |
| 14GT77 | Lacecap | 3 | 8 |
| Blue Sky | Lacecap | 2 | 7 |
| BM-1 (mutant of Blue Sky) | Mophead | 2 | 7 |
| Chikushinokaze | Lacecap | 3 | 3 |
| Corsage | Lacecap | 3 | 6 |
| Dance Party | Lacecap | 3 | 4 |
| Fairy Eye | Lacecap | 3 | 8 |
| HK01 | Lacecap | 4 | 4 |
| HK02 | Lacecap | 5 | 8 |
| Izunohana | Lacecap | 3 | 5 |
| Jogasaki | Lacecap | 3 | 8 |
| Kirakiraboshi | Lacecap | 4 | 4 |
| Nijima-1 | Lacecap | 1 | 4 |
| Posy Bouquet Kaycey | Lacecap | 3 | 8 |
| Posy Bouquet Grace | Lacecap | 3 | 8 |
| Sumidanohanabi | Lacecap | 4 | 5 |
| SK-1 | Lacecap | 2 | 8 |
| SKM-1 (mutant of SK-1) | Mophead | 2 | 8 |
- —Japan Society for the Promotion of Science and Grants-in-Aid for Scientific Research, KAKENHI
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Photosynthetic Processes and Mechanisms · Plant Genetic and Mutation Studies
Introduction
Most Hydrangea species have inflorescences composed of two types of flowers: decorative flowers with showy floral organs and plain nondecorative flowers. The sepals of decorative flowers are large and white, blue, pink, purple, and red, whereas those of nondecorative flowers are small and green. Decorative and nondecorative flowers of Hydrangea macrophylla differ in the shape of their sepals, number of floral organs, position of flowers on the inflorescence axes, and pedicel morphology [1, 2]. In contrast, the petal shapes are similar in both types of flowers, as are the stamens and pistils, which produce viable pollen and functional ovules, respectively. Most species in the genus Hydrangea bear corymb. In the corymb of Hydrangea species, the internodes on the main inflorescence axis are alternately short and long. In the corymbs of H. macrophylla and H. serrata, two decorative flowers are borne on each of the secondary inflorescences formed in pairs on the first and second nodes of the main inflorescence axis [3]. These four secondary inflorescences are located at the periphery of the corymb; therefore, the decorative flowers are also located at the periphery of the corymb, resulting in a frame-shaped phenotype. In the inflorescences of H. macrophylla and H. serrata, the frame-shaped phenotype is referred to as the lacecap phenotype. Furthermore, in these species, as in several other Hydrangea species, a mophead phenotype has been found, with many decorative flowers in the inflorescence. In the mophead phenotype, many decorative flowers with long pedicels completely overshadow nondecorative flowers with short pedicels, resulting in a globular inflorescence. This phenotype is found very rarely in wild populations of H. macrophylla or H. serrata; however, its discovery has led to the widespread use of these species as horticultural plants. Although this phenotype is a valuable trait in hydrangeas, the genes controlling it have not yet been elucidated.
The mophead phenotype of H. macrophylla is a recessive trait that appears when the mophead allele is homozygous or when different mophead alleles are combined [4, 5]. From the latter half of the 19th century to the beginning of the 20th century, three mophead cultivars of H. macrophylla from Japan and one from China were introduced into Europe and the USA [6]. Many mophead cultivars have been bred using these four germplasm lines and their descendants. Therefore, the number of mophead alleles utilized for breeding H. macrophylla is limited.
Uemachi et al. [2] compared the structure and developmental processes of the inflorescences of lacecap and mophead hydrangeas using a lacecap cultivar and mophead phenotype mutant (bud sport). The total number of flowers constituting the inflorescence decreased significantly because of the mutation from the lacecap to the mophead phenotype. The percentage of decorative flowers among all flowers in the inflorescence was 3.7% for lacecap and 66.5% for mophead. The fundamental characteristics of the mophead inflorescence structure were similar to those of the lacecap. In both phenotypes, nondecorative flowers were set on top of the inflorescence axes as terminal flowers, and pairs of axillary higher order inflorescences were set racemosely on most nodes of the axes. The only difference between the lacecap and mophead inflorescences was the structure of the upper nodes near the terminal flowers. Axillary higher order inflorescences were set on the upper nodes of the lacecap, whereas decorative flowers were set at comparable positions on the mophead. These results indicated that the mutation from the lacecap phenotype to the mophead phenotype was induced by the replacement of axillary higher order inflorescences with decorative flowers (Fig. 1). In other words, the mutation from lacecap to mophead causes the meristem identity of an axillary bud to switch from an inflorescence to a flower with large sepals and a long pedicel. Candidate genes that control both meristem identity and sepal morphology in Arabidopsis include APETALA1, a class A gene, and SEPALLATA class E genes [7–10]. These genes could contribute to inflorescence phenotype and sepal morphology in hydrangea. The development of DNA markers linked to the INFLORESCENCE TYPE (INF) locus has been reported. Although tightly linked INF markers have been developed, the causative gene for the mophead phenotype has not yet been identified. Waki et al. [5] constructed an SSR marker-based genetic linkage map and found that HS071 was linked to the mophead phenotype. Tränkner et al. [11] performed bulked segregant analysis using genotyping by sequencing and developed the mophead phenotype-linked markers, A109A110 and A133A134. According to the A109A110 marker-amplified sequences, two of the six alleles were linked to the recessive mophead allele. Wu et al. [12] performed a genome-wide association study (GWAS) and developed a cleaved amplified polymorphic sequence (CAPS) marker, Hy_CAPS_Inflo. Causative gene for INF should be around these markers.
Changes in inflorescence structure associated with mutation from the lacecap phenotype to the mophead phenotype [2]. The mutation from the lacecap phenotype to the mophead phenotype is induced by the replacement of axillary higher order inflorescences with decorative flowers.
The genome of H. macrophylla has been reported [13, 14]. One genome sequence is Hma_r1.2 sequence, which derived from ‘Aogashima-1’ introduced from its habitat in Aogashima Island [13]. The genome assembly consisted of 3779 scaffolds with a 2.2 Gb length, with a total length of 1078 Mb of 18 pseudomolecules, approximately half the length of the total genome sequence. A total of 36 930 genes were identified. Latest genome assembly is Hmc_EndlessSummerp1.0 sequence, which sequenced ‘Endless Summer’ [14]. The genome assembly consisted of 396 contigs (2.22 Gb length) that were assembled and scaffolded into the expected 18 pseudochromosomes. Utilizing genome sequencing and resequencing comparisons between alleles would contribute to clarifying the genes controlling inflorescence type and the mutations causing the mophead phenotype. Previously, LEAFY homologous genes and their mutations were identified as candidate causes of the double flower phenotype in H. macrophylla using genome sequence information and resequencing data [13]. Resequencing using short-read technology is an effective method for identifying causative mutations, particularly functional mutations caused by SNP or short INDEL. Recently developed advanced long-read technologies are powerful tools for identifying large insertions and deletions. Using long-read technologies, structural variants, such as a 10-kb retrotransposon insertion in maize and a 20-kb inverted repeat insertion in pineapple, have been detected [15, 16]. Therefore, adopting long-read technology for resequencing could effectively detect structural variant mutant alleles in hydrangea.
In this study, we aimed to identify the genes controlling the inflorescence type and the mutations causing the mophead phenotype. In this study, we identified SNPs that co-segregated with the inflorescence phenotype in a GWAS using F_2_ populations of H. macrophylla. The scaffold containing the SEP homolog locus, which was differentially expressed according to the inflorescence phenotype, coincided with the scaffold containing a marker that was strongly linked to the inflorescence phenotype. Therefore, the genome sequences of the SEP homolog, named TEMARY, were analyzed in several H. macrophylla cultivars. The results revealed that the H. macrophylla cultivars have three types of loss-of-function alleles for the TEMARY locus, and that the mophead cultivars carry only loss-of-function alleles.
Results
Prediction of the candidate gene at the INF locus
A GWAS and a database search for previously developed INF markers were conducted to identify the INF locus in the population. For the GWAS analysis, ‘Posy Bouquet Grace’ (lacecap), ‘Blue Picotee Manaslu’ (mophead), and their F_1_ individual 12GM1 (lacecap) and selfed population of 12GM1were used. The sample included 104 lacecap and 29 mophead phenotypes, which was not deviated from the Mendelian segregation ratio of 3:1 with Chi-square test (P = 0.394). ddRAD-Seq data were mapped to the Hmc_EndlessSummer_p1.0 genome [14]. As a result of GWAS, 160 of 11 404 SNPs were found to be significant. All significant SNPs (−log10(P-value) > 5.35) were located on 90.3 to 129.1 Mb on chromosome 04 (Fig. 2A and B). The highest -log10 (P-value) was detected for SNP 12GM1–02464 (pos: 117620, 023 on chromosome 04) (Fig. 2B). Genomic regions supported by SNPs with more than −20 of -log10(P-value) values from the highest SNP are listed in Supplemental Table S1. Although no SNP completely corresponded to the genotype and phenotype, SNP 12GM1–02445, 12GM1–02463, and 12GM1–02464 showed a lower number of lacecap phenotypes with homozygous reference alleles and the highest number of mophead phenotypes with homozygous reference alleles. These SNPs supported the region around 116 703 016 to 118 954 750, which was expected to be the INF gene-locating region. Previously developed tightly linked INF markers [11, 12] were mapped from 116 278 775 to 118 423 698 bp (Fig. 2C, Supplemental Table S2). This region overlapped with the region suggested by the GWAS results.
INF locus prediction using GWAS of F2 population 12GM1. (A) Manhattan plot on whole genome sequence for INF phenotype. (B) Manhattan plot on CM066985.1 (chromosome 04) sequence for INF phenotype. (C) Detailed INF locus around SNP 12GM1-02464. Nucleotide numbers correspond to Hmc_EndlessSummer_p1.0 genome. Corresponding scaffolds from Hma_r1.2 [13] are shown as blue arrows. White arrows indicate INF markers amplifying region and highest peak SNP12GM1-02464. The green arrow indicates candidate INF gene TEMARY.
Then, candidate causative genes of the INF phenotype in this region were searched. Because gene sequences were not provided for Hmc_EndlessSummer_p1.0 [14], the gene annotation from Hma_r1.2_1 [13] was used. Around this region, scaffolds 0060F, 0744F and 0340F from Hma_r1.2_1 genome were corresponded to 114–120 Mb region of chr04 of Hmc_EndlessSummer_p1.0 genomic sequence (Fig. 2C). In these scaffolds, 138 genes were predicted (Supplemental Table S3). To screen candidate causative gene by mRNA expression level, comparison between lacecap accessions (‘Blue Sky’ and SK-1) and their mophead sport mutant accessions (BM-1 and SKM-1, cuttings of the mutated branches) was conducted. Then, nine genes were suggested as differentially expressed (three times higher or lower in mutant accessions, FPKM in both original accessions were more than 0) (Table 1). Hma1.2p1_0060F.1_g032930 was a previously predicted candidate gene for mophead phenotype, since Arabidopsis homologous gene CYP97A5 (At1g13710), which was also similar with CYP78A10 (At1g74110), was suggested as floral organ development-related gene [14, 17]. However, it did not show expression difference in mutant accessions. Among differentially expression genes, Hma1.2p1_0060F.1_g032780 and Hma1.2p1_0060F.1_g033600 were suggested as homologues of morphology related genes. Hma1.2p1_0060F.1_g033600 had similarity with MYB106, which was suggested as cuticular pattern formation and trichome branching in Arabidopsis thaliana [18, 19]. According to Arabidopsis homologous gene MYB106, this gene was suggested not to contribute shoot morphologies. In addition, Hma1.2p1_0060F.1_g033600 showed scarce expression also in original accessions. These data did not positively support the possibility that Hma1.2p1_0060F.1_g033600 is a candidate gene for inflorescence phenotype. Hma1.2p1_0060F.1_g032780 is a partially predicted sequence of the candidate Sepallata sequence. This gene was predicted as candidate causative gene for the INF phenotype. As mentioned in the introduction, SEP is a gene that affects inflorescence structure and sepal morphology in Arabidopsis, and the SEP homologue is a strong candidate gene for controlling inflorescence phenotype in hydrangea. Here, we name this candidate gene ‘TEMARY’, which means mophead phenotype in Japanese.
Phylogenetic analysis based on putative amino acid sequences of SEPALLATA (SEP) homologous isoforms of H. macrophylla and SEP homologs of H. quercifolia, A. thaliana, G. hybrida, S. lycopersicum, V. vinifera, and P. hybrida. There were a total of 295 positions in the final dataset. The maximum likelihood method based on the JTT model [20] with a discrete gamma distribution (+G, five categories) was applied for phylogenetic analysis using the MEGA 11 software [21]. The tree with the highest log likelihood was shown. The maximum likelihood tree had a log likelihood of −8179.06 and + G parameter of 0.7609. The tree was rooted using AGL6 and AGL13 as an outgroup. The bootstrap values (1000 replicates) greater than 50% are indicated at the nodes. TEMARY is marked with a star.
Schematic model of eight TEMARY alleles. Varieties that included each allele sequence in long-read sequence were shown. Insertions >1.0 kb and loss of function mutations compared to allele 1 are shown. Green boxes indicate coding sequences. The red box and red characters indicate mutations that induce functional disruption. L: lacecap variety, M: mophead variety.
Identification and classification of TEMARY and SEP genes
TEMARY was suggested as partially predicted SEP family gene. To obtain full-length TEMARY transcript sequence and other SEP family genes, Iso-seq analysis was conducted. Then, fourteen SEP isoforms were identified. Fourteen SEP homologous isoforms were deduced to be transcripts from six loci (Supplemental Table S4 and Fig. 3). Multiple (2-4) isoforms were present at four loci. All isoforms were alleles possessed by the three samples (‘Blue Sky’, ‘Jogasaki’, Nijima-1) for Iso-Seq analysis. Of the six SEP homologous loci, two belonged to the SEP3 clade and four to the LOFSEP clade (Fig. 3). Among the homologous genes of the LOFSEP clade, TEMARY and another belonged to the FBP9/23 subclade, one belonged to the SEP1/2 subclade, and one belonged to the SEP4 subclade. TEMARY genomic sequence was coded on an 11 507–16 705 bp with eight exons in the hydrangea genome sequences according to mapped iso-seq sequence (Supplemental Table S5).
TEMARY allelic sequence identification and comparison
Because the lacecap phenotype is dominant, we hypothesized that TEMARY is functional in the dominant allele and disrupted in the recessive allele if TEMARY is the causative gene for the INF phenotype. Therefore, we identified and compared TEMARY allele sequences. Resequencing of cultivars with long-read technologies identified eight alleles (Alleles 1 to 8) with 10.4 to 17.3 kb of gene lengths (Fig. 4, Supplemental Table S6). Among them, alleles 1, 2, 3, 4, and 5 were suggested to be functional alleles because their deduced amino acid sequences produced the expected full-length protein (Fig. 5), and RNA-seq data for each allele were mapped on the expected exon region with a certain amount of mRNA expression (Supplemental Fig. S1).
In contrast, three alleles (Allele 6, 7, and 8) were functionally disrupted. For allele 6, a 7-kb insertion occurred in the third intron, with abnormal splicing (Figs 4, 6, Supplemental Fig. S1). Allele 7 possessed a nonsense mutation in the first exon, resulted in premature stop codon with only 17 amino acid residues (Fig. 5, Supplemental Fig. S2). Allele 8 possessed an 4.8-kb insertion in the first exon, with very little or no TEMARY mRNA (Supplemental Fig. S1). According to tree clustering derived from the genomic sequence alignment from the start codon to the stop codon, allele 6 branched from allele 1, allele 7 from allele 4, and allele 8 from allele 5 (Supplemental Fig. S3). These alleles were occurred independently in their history.
In the resequenced varieties, possessing functional allele (alleles 1 to 5) led to lacecap phenotype, and possessing only disrupted alleles (alleles 6, 7, and 8) led to mophead phenotype. Haplotype genome of mophead variety ‘Endless Summer’ included alleles 7 and 8 in their haplotype sequence [14], and lacecap accessions ‘Aogashima-1’ genome sequence possessed allele 1 [13] (Supplemental Table S5), published genome sequences also showed coincidence with above rule.
According to allelic sequences, it was clarified that ‘Blue Sky’ and BM-1 possess alleles 2 and 7, and SK-1, and SKM-1 possess alleles 2 and 8 at the TEMARY locus. If TEMARY gene is the causal gene for these sport mutants, RNA reads of functional allele 2 should be decreased or lost in sport mutants. In the mapping analysis, reduced expression levels at 0.15 to 0.18 times of the functional TEMARY allele 2 were observed in the sport mutants. (Table 2). In both SK-1 and SKM-1, scarce expression levels were observed on allele 8 (Supplemental Fig. S4). For ‘Blue Sky’ and BM-1, while allele 7 FPKM were similar level, allele 2 was largely decreased in BM-1 (Table 2). In BM-1, RNA mapping of exons 3 to 6 was scarcely observed (Supplemental Fig. S5). The results of expression analysis by qRT-PCR targeting all TEMARY alleles supported the results of the RNA-seq analysis (Fig. 7). The expression levels of the TEMARY gene in the sport mophead mutants were reduced compared to the original lacecap cultivars, suggesting that the difference was due to differences in the expression of allele 2.
Deduced amino acid sequence alignment of TEMARY alleles. Alleles with red character indicate functionally disrupted alleles.
RNA-Seq mapping of TEMARY allele 1 in ‘Niijima’ (allele 1/4) and allele 6 in ‘Honajisai’ (allele 6/8). Green boxes indicate expected coding sequences. Gray graphs show coverage tracks, and red curves show splice junction tracks obtained by the Integrated Genome Viewer.
Comparison of expression levels of TEMARY between the original lacecap cultivars (Blue Sky, SK-1) and their sport mophead mutants (BM-1, SKM-1). Exons 1 and 4 of all TEMARY alleles were amplified. Inflorescence buds with a diameter of 3–5 mm were used for analysis. HmActin was used as an internal control. qRT-PCR analysis was performed in three biological replicates. Vertical bars indicate SE (n = 3). * and ** indicate significant differences between the original cultivar and its sport mutant at P < 0.05 and 0.01, respectively, by T-test.
Development and application of the TEMARY markers
According to each allelic sequence, DNA markers for discriminating TEMARY alleles were developed as Temary-1, Temary-2, Temary-3, and Temary-4 (Supplemental Table S7, Supplemental Fig. S6). The combined use of Temary-2 and Temary-1 distinguished between alleles 1 and 8. According to the genotypes of the accessions, the mophead phenotype was observed when possessing only functionally disrupted alleles 6, 7, and 8, with the exception of sport mutants BM-1 and SKM-1 (Table 3). To detect causative mutations for alleles 6, 7, and 8, using agarose gel electrophoresis-based methods, Temary-3 and Temary-4 markers were additionally designed. The dCAPS marker, Temary-3, was designed to distinguish between the presence and absence of nonsense mutations in the first exon. The Temary-4 marker was designed to distinguish between the presence and absence of large insertions in the first exon. Note that the Temary-1 marker (with or without the 6-FAM label) easily distinguished between allele 6-derived PCR fragments by agarose gel electrophoresis. These markers can be used to distinguish between mutations and functional alleles.
Coupling of alleles between INF markers and TEMARY
Coupling of TEMARY alleles, SNP 12GM1–02464, and reported alleles of INF markers [11, 12] was performed (Supplemental Table S8). For SNP 12GM1–02464, the mophead-linked allele G was also linked to the lacecap-linked TEMARY alleles 1, 4, and 5. However, the genotype of the F_1_ individual 12GM1 was 3/8, which was coupled with each phenotype-linked allele in 12GM1–02464. Therefore, the phenotype and genotype were highly associated in the selfed F_2_ population. In contrast, six individuals did not cosegregate with the phenotype and genotype. Although phenotyping or handling errors were possible, unexpected cross-pollination of pollen with alleles 1, 4, and 5 of TEMARY was considerable.
Discussion
TEMARY as a gene responsible for the inflorescence phenotype
In expression and linkage analyses, the homeobox SEP homologous gene TEMARY was identified as a candidate gene for the mophead phenotype in H. macrophylla. TEMARY is located at the 117 Mb position on chromosome 04, where previously developed INF markers are located in close proximity [11, 12]. The presence or absence of the TEMARY function in each allele was coupled with a phenotype; the lacecap phenotype was observed in the presence of a functional allele, and the mophead phenotype was observed in the absence of a functional allele. These data strongly support the hypothesis that TEMARY is responsible for the mophead phenotype.
Recently, Wu et al. [14] suggested the possibility of CYP78A5, which was corresponded to Hma1.2p1_0060F.1_g032930 in Hma_r1.2 genome, as candidate gene for inflorescence shape and showed that the genotype of this gene can explain the phenotypes of the mophead cultivar ‘Endless Summer’ and the lacecap cultivar ‘Veitchii’. This gene was discovered based on its putative function in several genes located near a marker as obtained through linkage analysis. However, TEMARY, which is located near this gene, was not identified, even though it is the most likely candidate based on its putative function.
It was revealed that the phenotypes of 27 mophead cultivars could be explained by three types of loss-of-function TEMARY alleles. The pedigree in these samples were also in accordance with expected TEMARY genotype. ‘Chikushinomai’ was bred by Fukuoka Agriculture and Forestry Research Center, Japan (https://www.farc.pref.fukuoka.jp/hinshu/hinshu.html, In Japanese). ‘Chikushinomai’ (genotype 7/8) is the progeny of mophead accession ‘Posy Bouquet Grace’ (3/8) and lacecap 03JP1 (3/7), and 03JP1 is the progeny of ‘Jogasaki’ (3/8) and Paris (7/8). In addition to selfed population of 12GM1, we confirmed that selfed 14GT77 (3/8) (progeny of ‘Posy Bouquet Grace’ (3/8) × ‘Chibori’ (8/8)) also not deviated from the Mendelian segregation ratio of 3:1 with Chi-square test (P = 0.37), which included 55 lacecap and 14 mophead phenotypes. These results showed that TEMARY genotype was well coupled with inflorescence phenotype.
The result that the mophead phenotype can be explained by three loss-of-function alleles rather than a single allele or DNA marker strongly supports that TEMARY is the causal gene for the inflorescence phenotype, since the probability of this being a coincidence is much lower. Furthermore, analysis using two sets of sports mutant lines also suggested that new loss-of-function mutations, which were not present in the original cultivar, occurred in the TEMARY allele 2 of the sports mutants. These results suggested that TEMARY was responsible for the inflorescence phenotype.
Loss-of-function TEMARY alleles possessed by the H. macrophylla cultivars
Many H. macrophylla cultivars have been bred by crossbreeding four mophead cultivars introduced in Europe and the USA between the latter half of the 19th century and the beginning of the 20th century. Thus, it was estimated that there were only a few types of mophead alleles in the H. macrophylla cultivars. In this study, three loss-of-function TEMARY alleles were identified that could explain the phenotypes of the mophead cultivars. Furthermore, these three loss-of-function TEMARY alleles coupled with previously developed INF markers co-segregated with the phenotypes of the accessions provided in each study [11, 12]. These results suggest that there are three main mophead alleles in H. macrophylla cultivars, all of which are loss-of-function TEMARY alleles. Although it has been suggested that the introduction of further alleles through recent mutations in cultivars occurs quite frequently in production areas [22]. In addition, sports mutants BM-1 and SKM-1 possibly possess new mutant alleles in allele 2. Therefore, three alleles are not all mophead alleles in the whole population. Even if new mophead genetic resources are used for breeding in the future, genetic markers for inflorescence phenotypes can be easily obtained by analyzing the sequences of their TEMARY genes.
Although we developed a Temary-1 marker, PCR and subsequent fragment analysis were required. Therefore, simpler and more cost-effective markers are needed for breeding programs. Three loss-of-function alleles were suggested to occur from different functional alleles (allele 6 from 1, 7 from 4, and 5 from 8) based on tree clustering of the TEMARY genomic sequence. Although ideal INF markers should simultaneously distinguish the three loss-of-function alleles from the functional alleles, such DNA polymorphisms are rare. We did not find such a polymorphism in genome sequence of TEMARY. In this respect, the Hy_CAPS_Inflo marker was suggested to be highly valuable as it could successfully distinguish phenotypes using only a single SNP.
There may be functional differences among the loss-of-function alleles. In H. macrophylla accessions bred in Japan before 20th century, such as ‘Honajisai’, ‘Uzuajisai’, and ‘Junihitoe’, allele 6 was included. However, recent cultivars bred in Japan rarely contain allele 6. Cultivars with genotype 6/6 were not observed. This might reflect the breeding selection history in Japan, consciously or unconsciously weeding out allele 6 according to other characteristics. Tränkner et al. [11] suggested that individuals with the B1 homozygous genotype (coupled with 7/7 in TEMARY) in the A109A110 marker frequently exhibited a nonflowering phenotype. Although it is not clear whether the nonflowering phenotype is caused by TEMARY, TEMARY studies may reveal relationships among TEMARY alleles, flowering phenotypes, and breeding selection in the future.
Comparison of TEMARY between the original cultivar and its sport mutant
To confirm that TEMARY is the causal gene for the mophead phenotype, we used two sets of original lacecap cultivars and mophead mutants; these were ‘Blue Sky’ and BM-1, SK-1, and SKM-1. The original lacecap cultivars have heterozygous functional and disrupted alleles, and expression levels of the functional allele (allele 2) were reduced in the mophead mutants. RNA-seq mapping and qRT-PCR results suggested the expression levels of most exons in allele 2 were reduced in both mophead mutants. These suggest that in allele 2 of each mutant, a novel DNA polymorphism occurred in a region containing multiple exons or in the promoter region. In BM-1, the expression level of the functional allele 2 was decreased, while the expression level of the disrupted allele 8 was maintained at a high level, similar to that in ‘Blue Sky’. This result suggests that the decreased expression of allele 2 is not due to mutation of the upstream gene but due to mutation of allele 2. Future identification of such mutations should reveal the mechanisms for reduced expression of TEMARY allele 2 and would provide direct evidence for the idea that TEMARY gene function necessity for lacecap phenotype formation.
The presumed function of TEMARY belonging to the LOFSEP clades
In the process of inflorescence development in H. macrophylla, the only difference between the inflorescences of lacecap and mophead is the differentiation of axillary primordia at the upper nodes near the terminal flowers in primary and axillary higher order inflorescences [2]. These primordia differentiate into axillary higher order inflorescences in the lacecap and decorative flowers in the mophead. Thus, Uemachi et al. [2] predicted that the genes that determine the inflorescence phenotype of H. macrophylla are likely to be involved in inflorescence determinacy. TEMARY is one of the SEP homologous genes that may be involved in inflorescence determinacy. A. thaliana has four SEP genes, of which SEP3 plays a leading role in flower organogenesis as one of the ABCE genes [8, 23–25]. Members of the SEPALLATA (SEP) MADS-box subfamily in angiosperms are divided into two major sister clades: the SEP3 and LOFSEP clades [26, 27]. The LOFSEP clade is functionally more diverse than the SEP3 clade and its functions include controlling inflorescence determinacy and shape [28]. TEMARY belongs to the FBP9/23 subclade, which is one of the three subclades of the LOFSEP clade (Fig. 3). The FBP9/23 subclade includes JOINTLESS-2 and ENHANCER-OF-JOINTLESS-2 (J2 and EJ2) of tomatoes (Solanum lycopersicum), which are involved in inflorescence determination [29]. J2 and EJ2 repress inflorescence branching; therefore, their loss-of-function mutants produce inflorescences with more flowers. The SEP gene of rice (Oryza sativa) OsMADS34/PANICLE PHYTOMER2 (PAP2) and SEP homologs of Gerbera hybrida have similar functions to J2 and EJ2 of tomato [30–33]. However, the presumed function of TEMARY is opposite to that of these SEP homologous genes. The change in inflorescence structure, presumably caused by the functional deletion of TEMARY is the replacement of axillary higher order inflorescences with decorative flowers and the induction of inflorescences with fewer flowers [2]. In the phylogenetic tree, another SEP homolog in H. macrophylla in addition to TEMARY belonged to the FBP9/23 subclade (Fig. 3). Similarly, two SEP homologous genes in H. quercifolia belonged to the FBP9/23 subclade, forming a cluster with two SEP homologous genes in H. macrophylla. Gene duplication causes diversification of gene function [34]. In the genus Hydrangea, unique functional differentiation may have occurred after the duplication of an SEP homologous gene belonging to the FBP9/23 subclade. In the future, the involvement of TEMARY in inflorescence determination should be examined together with another paralog belonging to the same FBP9/23 subclade as TEMARY (Fig. 3). However, the loss of function of tomato EJ2 and rice PAP2 resulted in enlarged sepals and lemmas, respectively, which is consistent with the presumed function of TEMARY. This suggests that, like the LOFSEP genes in rice and tomato, Temary retains the ability to control the size of outer floral organs.
Conclusion and future perspectives
The results of this study suggest that TEMARY is responsible for the inflorescence phenotype. In the genus Hydrangea, species such as H. serrata, H. involucrata, and H. arborescens have a clustered flowering phenotype similar to that of H. macrophylla. Analysis of TEMARY homologs will be an important approach for research on the mechanisms of inflorescence phenotypes in these species and for the development of markers. In H. quercifolia and H. paniculate, there is also a phenotype in which many decorative flowers are born in the panicle, which is an important target for breeding. The molecular mechanism underlying this phenotype is unclear. The phylogenetic tree (Fig. FPKM Accession Phenotype Allele 2 Allele 7 Allele 8 Blue Sky Lacecap 13.59 12.35 - BM-1 (sport mutant of BlueSky) Mophead 2.39 9.51 - SK-1 Lacecap 17.60 - 0.11 SKM-1 (sport mutant of SK-1) Mophead 2.72 - 0.14 ) shows that two genes belonging to the FBP9/23 subclade were present in H. macrophylla, and their respective orthologous genes were also present in H. quercifolia. This suggests that the functions of TEMARY are conserved in other Hydrangea species. The relationship between the TEMARY ortholog and the inflorescence phenotype of H. quercifolia should be investigated in the future.
Methods
RNA-Seq analysis and acquisition of contigs and isoforms
RNA samples from each cultivar were obtained from potted plants grown in greenhouses at the University of Shiga Prefecture, Japan. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Hilden, Germany). The RNA concentration and integrity were measured using a NanoDrop spectrophotometer and an Agilent 2100 Bioanalyzer. The extracted total RNA was treated with oligo (dT) magnetic beads to isolate the mRNA, and the fragmented mRNA was used to construct cDNA. The cDNA fragments were amplified using the PCR primers. Amplified cDNA libraries were sequenced using Illumina HiSeq 2000 for paired–end–sequencing (2 × 100 bp) of SK-1 and SKM-1 samples, Illumina HiSeq 2500 for paired–end–sequencing (2 × 100 bp) of ‘Blue Sky’ and BM-1 samples, Illumina NovaSeq 6000 for paired–end–sequencing (2 × 150 bp) of ‘Honajisai’ samples, and Roche 454 GS FLX+ for single–end–sequencing (600–700 bp) of mixed RNA samples of ‘Blue Sky’ and BM-1. The reads from inflorescence buds (3–17 mm in diameter) of ‘Blue Sky’ and BM-1 using Illumina HiSeq 2500 (‘Blue Sky’: 55328676 reads, total 5588 Mbp, BM-1: 53077204 reads, total 5361 Mbp), and Roche 454 GS FLX+ (‘Blue Sky’: 55328676 reads, total 5588 Mbp, BM-1: 53077204 reads, total 5361 Mbp) were added to the RNA-Seq reads from the H. macrophylla leaf and flower available in the database (SRR610319: 785504 read, total 282 Mbp, SRR68791: 359683 bp, total 36 Mbp) and were assembled using a GS de novo Assembler version 2.6 (Roche Diagnostics) with parameters of minimum overlap length of 40 bp and minimum overlap identity of 90%.
The mixed RNA sample from inflorescence buds, leaves, decorative flowers, and nondecorative flowers of ‘Blue Sky’, ‘Jogasaki’ and a wild individual of H. macrophylla was reverse-transcribed using the SMARTer™ PCR cDNA Synthesis Kit (Clontech, USA). After determining the optimal number of PCR amplification cycles, cDNA was amplified using PrimeSTAR GXL DNA Polymerase (Clontech). After purification with AMPure PB Beads, the cDNA products were used to construct SMRTbell template libraries using SMRTbell Template Prep Kit 1.0 (PacBio). Sequencing was performed using the PacBio Sequel platform. Raw reads were processed into circular consensus sequence reads and subsequently clustered to consensus isoforms by Iso-Seq3 package included in PacBio SMRT Link (https://github.com/PacificBiosciences/IsoSeq).
Phylogenetic analysis of homologous isoforms of SEP
Using RNA-Seq data and reads from leaves and flowers of H. macrophylla available in the database, 29 272 EST contigs were constructed. A BLAST search was performed on these contigs, and nine SEP homologous contigs were obtained (Supplemental Data S1). These contig sequences were used as query sequence for identification of the iso-seq sequence of SEP homologs. The SEP family genes of A. thaliana, Vitis vinifera, S. lycopersicum, Petunia hybrida, and Gerbera hybrida were used to create phylogenetic trees, referring to the work of Dreni and Ferrándiz [28] on the evolution of the SEP subfamily. The deduced amino acid sequences were aligned using the CLUSTAL W multiple sequence alignment algorithm, and the maximum likelihood method based on the JTT model [20] with a discrete gamma distribution (+G, five categories) was applied for phylogenetic analysis using MEGA 11 software [21]. In both phylogenetic analyses, bootstrap tests were performed with 1000 replicates.
Prediction of the genomic locus for the mophead phenotype
To detect SNPs linked to the mophead phenotype, ddRAD-Seq analysis was performed on the 12GM1 F_2_ population [13] and their parent ‘Posy Bouquet Grace’ (lacecap), ‘Blue Picotee Manaslu’ (mophead) and their F_1_ individual 12GM1. An F_1_ individual, 12GM1, was obtained by crossing parents and then, 12GM1 was selfed to obtain F_2_ individuals. The inflorescences used for cross- or self-pollination were covered with double nonwoven fabric to prevent cross-contamination.
The low-quality ddRAD-Seq sequences were removed, and adapters were trimmed using PRINSEQ (−trim_right 1 -trim_qual_right 10 -min_len 100 -derep) and fastx_clipper (−a AGATCGGAAGAGC) in the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit; version 0.10.1). The filtered sequences were mapped onto Hmc_EndlessSummer_p1.0 [14] using Bowtie2 [35]. SNP calling was performed using BCFtools [36], and high-quality SNPs were selected by filtering conditions with a minimum read depth of 5 (−-min DP 5), a minimum SNP quality of 10 (−-minQ 10), minor allele frequency greater than 0.05 (−-maf .05), a maximum proportion of missing data of 10% (−-max-missing 0.9), and remove indels (−-remove-indels) using VCFtools [27].
GWAS analysis was performed using TASSEL 5 [38], and the GLM model was used to detect significantly associated loci. Genotypes and phenotypes were used in the GLM model, with the first five components of the PCA as the population structure matrix. The threshold values for associated SNPs were obtained by the Bonferroni correction, which was calculated as follows: −Log10(P) ≥ −Log10(0.05/11404) = 5.35.
Previously developed INF marker regions were searched in the genomic sequences of H. macrophylla. Each primer sequence for INF markers [5, 11, 12] was searched using BLAST.
Gene annotation of the INF locus was added using the Hma_r1.2_1 genome [13]. The corresponding gene locus for Hmc_EndlessSummer_p1.0 was searched using BLAST.
Resequencing of H. macrophylla accessions and identification of the TEMARY allele sequence
Resequencing of H. macrophylla accessions ‘Honajisai’ (mophead), ‘Blue Picotee Manaslu’ (mophead), ‘Frau Yoshimi’ (mophead), ‘Blue Sky’ (lacecap), ‘Sumidanohanabi’ (lacecap), and ‘Posy Bouquet Grace’ (lacecap) was performed using a Sequel System (PacBio, Menlo Park, CA, USA). An SMRT library was constructed using the SMRTbell Express Template Prep Kit 2.0 (PacBio, Menlo Park, CA, USA) according to the manufacturer’s protocol and sequenced using SMRT Cell 8 M on a Sequel System II to obtain high-fidelity (HiFi) long reads.
Obtained sequences of each accession were converted into BLAST database using the ‘make blastdb’ function in CLC mainworkbench (Qiagen). The TEMARY genomic sequence in Hma_r1.2_1 [13] was used as a query, and hit sequences were extracted and aligned using the ‘create alignment’ function with the default settings, and the consensus sequence was obtained from the alignment. Raw sequence reads data of ‘Aogashima-1’ [13] was used for TEMARY allele sequence of ‘Aogashima-1’.
RNA-Seq mapping TEMARY alleles
To compare RNA mapping pattern among alleles, RNA-Seq sequences for ‘Nijima-1’ (1/4), ‘Blue Sky’ (2/7), ‘Jogasaki’ (3/3), ‘Sumidanohanabi’ (3/3), and ‘Honajisai’ (6/8) were used. TEMARY alleles 1–8 were used as references. RNA-Seq data were mapped using hisat2 with default parameters and no-discordant and no-mixed options [39]. The SAM files obtained were sorted and converted into BAM files using the same tools [36]. The obtained BAM data were visualized using Integrated Genomics Viewer [40].
For comparison of TEMARY mRNA mapping between the original lacecap cultivars and their mophead mutant, ‘Blue Sky’, BM-1, SK-1, and SKM-1 RNA-Seq were mapped for combined sequence with Aogashima-1 genome (Hma_r1.2_1) and TEMARY alleles; alleles 2 and 7 for ‘Blue Sky’ and BM-1, alleles 2 and 8 for SK-1 and SKM-1. RNA-Seq mapping and visualization were performed as described above. FPKM for each gene and TEMARY alleles were calculated by StringTie [41].
qRT-PCR analysis
Total RNA was reverse transcribed using ReverTra Ace (Toyobo Co., Ltd, Osaka, Japan) according to the manufacturer’s instructions. Gene expression levels were analyzed by qRT-PCR using the TB Green Pre-mix Ex- Taq II Fast qPCR (Takara-Bio, Kusatsu, Japan) with a Thermal Cycler Dice Real Time System III (Takara-Bio), according to the manufacturers’ instructions. A primer set for amplifying exon 1 was designed within exon 1 (Supplemental Table S9). For amplifying exon 4, a forward primer was designed at the boundary between exons 3 and 4, and a reverse primer was designed at the boundary between exons 4 and 5. qRT-PCR cycling was performed as follows: 95°C for 30 s, followed by 40 cycles at 95°C for 5 s and 60°C for 30 s. The comparative delta–delta Ct method was used to calculate expression levels [42]. HmActin was used as an internal control. qRT-PCR analysis was performed in three biological replicates.
TEMARY marker development and application for accessions
Alignment of TEMARY alleles was performed using the CLC Main Workbench (Qiagen). DNA markers were developed based on TEMARY allele alignment. To discriminate between the alleles, an FAM-labeled SSR marker (Temary-1) and an INDEL marker (Temary-2) were designed. Primers were selected by visual observation. The border of the causative inserted sequences that induced functional defects was used to design primers to develop a DNA marker for detecting allele 8 (Temary-3). For the detection of nonsense mutation on allele 7, the dCAPS marker (Temary-4) was designed.
PCR amplification was performed in a 10-μl reaction mixture containing 5 μl of 2x GoTaq Master Mix (Promega), 2.5 pmol of primers, and 5 ng of template DNA for all developed markers. The DNA samples were amplified under the following cycling conditions: for Temary-1, Temary-2, and Temary-4, 35 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 2 min, and a final extension of 5 min at 72°C. For Temary-3, 35 cycles of 94°C for 1 min, 58°C for 1 min, and 72°C for 2 min, and a final extension of 5 min at 72°C.
The PCR products were stained with 1x GRRED (Biocraft) and separated on a 2.5% (w/v) agarose gel in TAE buffer. For the Temary-4, a restriction enzyme assay was performed for the PCR product at 37°C for 3 h using NlaIII (New England Biolabs, Ipswich, MA, USA). The PCR products were stained with 1x GRRED (Biocraft) and separated on a 2.5% (w/v) agarose gel in TAE buffer. For the Temary-1 marker, the amplified PCR products were separated and detected using a PRISM 3130xl Genetic Analyzer (Applied Biosystems). The sizes of the amplified bands were scored against an internal standard DNA (400HD-ROX, Applied Biosystems, USA).
Supplementary Material
Web_Material_uhae332
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Uemachi T, Kato Y, Nishio T. Comparison of decorative and non-decorative flowers in Hydrangea macrophylla (Thunb.) Ser. Sci Hortic. 2004;102:325–34
- 2Uemachi T, Kurokawa M, Nishio T. Comparison of inflorescence composition and development in the lacecap and its sport, hortensia Hydrangea macrophylla (Thunb.) Ser. Hort J. 2006;75:154–60
- 3Uemachi T, Nishio T. Relationship between the inflorescence structure and setting of decorative flowers on lacecap cultivars of Hydrangea macrophylla (Thunb.) Ser. And H. Serrata (Thunb.) Ser. Hortic Res. 2005;4:435–8 (In Japanese with English abstract)
- 4Uemachi T, Okumura A. The inheritance of inflorescence types in Hydrangea macrophylla. Hort J. 2012;81:263–8
- 5Waki T, Kodama M, Akutsu M. et al. Development of DNA markers linked to double-flower and Hortensia traits in Hydrangea macrophylla (Thunb.) Ser. Hort J. 2018;87:264–73
- 6Haworth-Booth M . Hydrangeas. 5th ed. London: Constable; 1984:
- 7Bowman JL, Alvarez J, Weigel D. et al. Control of flower development in Arabidopsis thaliana by APETALA 1 and interacting genes. Development. 1993;119:721–43
- 8Ditta G, Pinyopich A, Robles P. et al. The SEP 4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr Biol. 2004;14:1935–4015530395 10.1016/j.cub.2004.10.028 · doi ↗ · pubmed ↗