Recurrent intussusception in a child revealing a neuroendocrine tumor within a Meckel's diverticulum: a case report
Hussein Naji, Reem Allateef, Aftab Ahmed, Shahzad Yousaf, Ghassan Nakib, Rawan Alhalabi

TL;DR
A young girl had multiple intussusception episodes, leading to the discovery of a rare neuroendocrine tumor in her Meckel's diverticulum.
Contribution
This is the first reported case of a pediatric neuroendocrine tumor within a Meckel's diverticulum.
Findings
A child with five intussusception episodes was found to have a low-grade neuroendocrine tumor in her Meckel's diverticulum.
The tumor showed no necrosis, suggesting a slow-growing and less aggressive nature.
The case highlights the need for prompt surgical removal of Meckel's diverticulum to detect and treat tumors early.
Abstract
Meckel's diverticulum (MD) is a common congenital anomaly of the gastrointestinal tract, often asymptomatic but occasionally presenting with various complications. Neuroendocrine tumors (NETs) are slow-growing neoplasms that mostly originate from the small intestine. The typical age for the presence of a carcinoid tumor within MD is above 50s. NETs reported to be the most common primary malignancy originating from MD in elderly. We report the first case of a female child who demonstrated five episodes of intussusception, ending in surgical resection revealing a NET within MD. The histopathology study proved a low-grade tumor with no evidence of necrosis. Current literature lacks definitive guidelines for managing NET in MD cases, especially in pediatric populations, necessitating further research for optimal treatment strategies. Prompt removal of the diverticulum upon discovery is…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
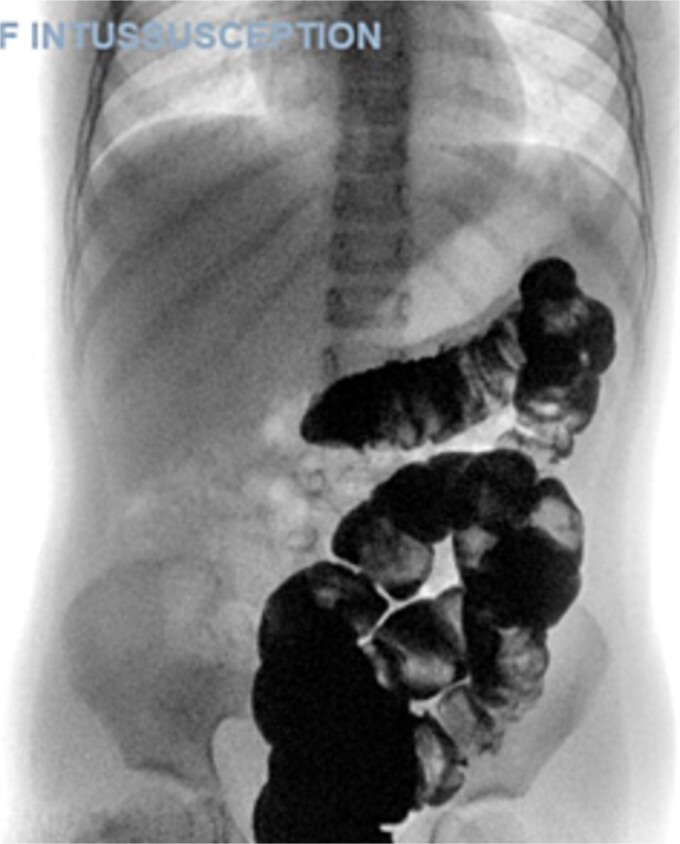 Figure 1
Figure 1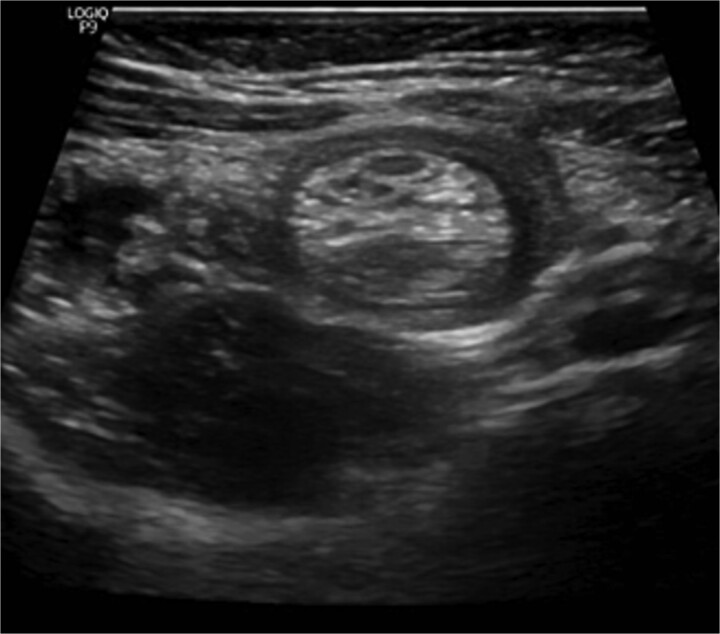 Figure 2
Figure 2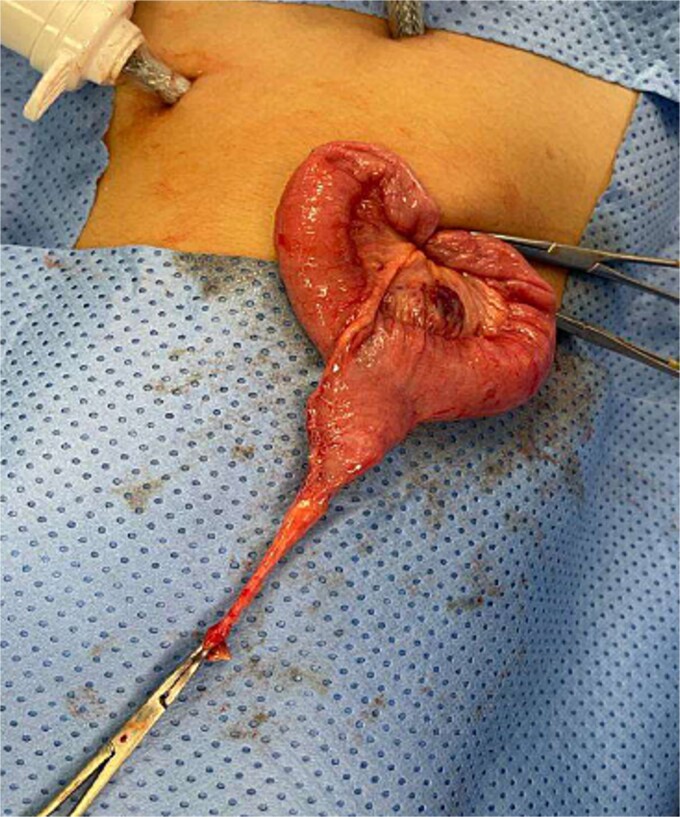 Figure 3
Figure 3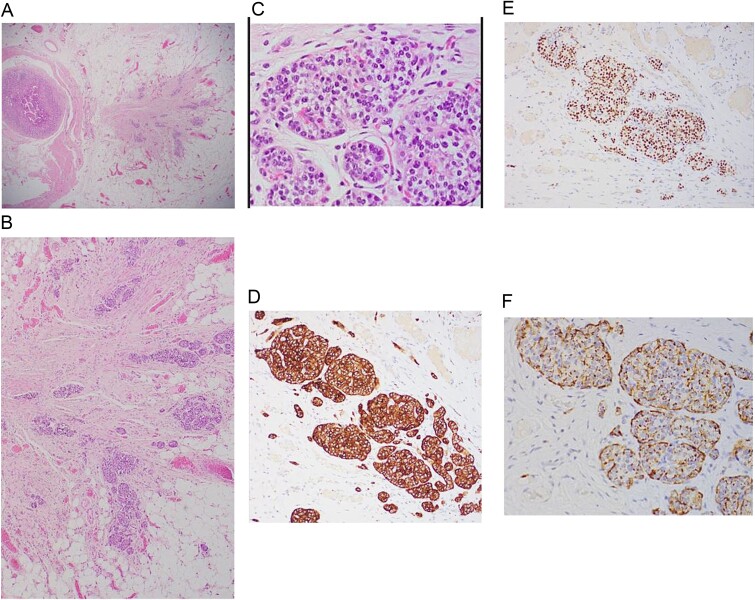 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGastrointestinal disorders and treatments · Gastrointestinal Tumor Research and Treatment · Tumors and Oncological Cases
Introduction
The omphalomesenteric duct is a canal that connects the midgut to the yolk sac in the fetus. Ideally involutes during the 5th-8th weeks of gestation, providing nutritional support until the formation of the placenta [1]. Meckel's diverticulum (MD) is an embryonic remnant pouch resulting from incomplete obliteration of the omphalomesenteric duct [1–3]. It is generally asymptomatic but may present with painless rectal bleeding, intussusception, diverticulitis, axial torsion, perforation, or peritonitis [1–3]. It is known to contain small intestinal tissue and gastric mucosa and has been documented in adults only to harbour tumors [1–4]. In this report, we present a novel case of recurrent intussusception in a child, which after surgical resection revealed a neuroendocrine tumor (NET) within a MD.
Case report
A 5-year-old female presented with a 1-day history of abdominal pain and vomiting. Physical examination revealed abdominal tenderness on the right side. The patient had a history of four previous episodes of intussusception, all of which were conservatively reduced with hydrostatic contrast enema (Fig. 1). The most recent episode occurred 3 months prior to the current presentation. An MRI enterography performed after the fourth episode showed no abnormalities.
Therapeutic enema for intussusception reduction (3rd attack).
The blood tests showed raised inflammatory markers. An abdominal ultrasound confirmed the presence of intussusception (Fig. 2). A decision was made for a laparoscopic reduction of the intussusception and inspecting for a leading point. A MD was demonstrated and resected accordingly (Fig. 3). The location of intussusception was at the ileo-cecal area. The affected bowel was viable. The position of the MD was in the terminal ileum and it was the lead point for the intussusception. No lymph node involvement. Also, a tumor located at the base of the MD ~4 mm from the resection margin, was found and resected.
Ultrasound of the abdomen showed a target sign of intussusception (5th attack).
Meckel’s diverticulum as a lead point for recurrent intussusception.
Histopathological assessment (Fig. 4) revealed small intestinal mucosa showing ectopic gastric tissue composed of foveolar and oxynetic glands, compatible with MD. In addition, a tumor was found. Composed of ribbons and cords of monomorphic cells with salt-pepper chromatin pattern and plenty of cytoplasm that show strong expression of Synaptophysin, INSMI, and Chromogranin A. Ki-67 proliferation index is low; <1% of tumor cells, compatible with NET grade 1. The tumor measured 4 mm, with no evidence of perineural or vascular invasion. Following the surgery, the child made a complete recovery and remained free of further episodes of intussusception during one year of follow-up in the clinic.
A: Low power magnification of H&E stain; tumor nest in the wall of the bowel, ribbons and cords of monomorphic cells with salt-pepper chromatin pattern, typically seen in endocrine tumors. B: Neuroendocrine tumor in the wall of bowel and fat (×20 magnification). C: Solid nest of tumor (high power magnification ×40). D: Immunohistochemistry Synaptophysin positive; Synaptophysin stains vesicles in neuroendocrine cells. E: Immunohistochemistry INSMI positive, a specific marker for neuroendocrine. F: Immunohistochemistry AE1/AE3 positive, a neuroendocrine marker.
Discussion
MD is usually lined by two types of mucosae: the innate intestinal mucosa and a heterotopic mucosa, with the gastric mucosa being a common heterotopic variant [5, 6]. Roughly, <15% of patients might exhibit extra tissue within the diverticulum, and the incidence of containing a primary tumor is as low as 0.5%, only reported in adults in the literature [1–5].
Carcinoid tumors, recently known as NETs, are slow-growing neoplasms arising from enterochromaffin cells with the ability to secrete a diversity of proteins and hormones. Most commonly originates from the small intestine, primarily the ileum [5–7].
Generally, ~80% of the affected people are symptomless. The known presentations are non-specific. Consequently, 50% of the patients will be misdiagnosed for at least 2–7 years, resulting in advanced stages of malignancy with up to 70% demonstrating metastasis [5–8].
The typical age for the presence of a carcinoid tumor within MD is 55 years, with a double occurrence in men compared to women [6, 8]. Modlin et al. stated that less than 1% of all carcinoids in adults appear in the MD [8].
Two papers published involved children. However, neither paper involved an incidental diagnosis of NET. The first article, published in 1969 [9, 10], included 40 cases, with only a patient of 12 years old with diverticulitis, not recurrent intussusceptions.
The second article, published in 2004, is an analysis of 13 715 cases of carcinoid tumors from 1950 to 1999. A 14-month-old child with hematochezia, reported in 1996 [8]. These studies illustrate the rarity of neuroendocrine tumors in MD, especially in children. Our case, which uniquely combines recurrent intussusceptions with a NET in MD, has not been previously published.
The Surveillance, Epidemiology, and End Results Program of the National Cancer Institute reported NETs as the most common primary malignancy originating from MD (76.5%), followed by adenocarcinoma (11.4%), GI stromal tumor (10.8%), and lymphoma (1.3%) [4–6].
The WHO issued a classification that categorizes these tumors as Grade 1 (G1-NET), Grade 2 (G2-NET), Grade 3 - neuroendocrine carcinoma (G3-NEC) large- or small-cell type, and mixed adenoneuroendocrine carcinoma. However, this grading is based on mitotic count besides using Ki-67 index. G1- NET is known to be composed of tumor cells dominating round or oval nuclei with salt-pepper chromatin and eosinophilic granular cytoplasm. G1-NET is concluded by a mitotic count of <2 per 2 mm^2^ (10 high power fields [HPF], 40× magnification) and/or ≤ 2% Ki-67 index. Mostly, the used stains are synaptophysin, chromogranin, CDX2 (identifying the origin, mainly reflects the intestine), PAX8 (positive in the majority of rectal and pancreatic), CD56 / NCAM1 (low specificity) and CEA [8–13].
In our case, synaptophysin, chromogranin A, and INSMI stains were positive, and Ki-67 index is <1%, leading to the diagnoses of G1-NET.
In adults, the treatment of MD-related NET typically follows staging guidelines. Regular exclusively MD surgical removal is considered satisfactory for lesions <10 mm [14–16], while substantial tumors may require resection of the ileal tract and corresponding mesentery. Metastatic lesions are managed with chemotherapy and symptomatic inhibition therapy [4, 17].
Although most literature supports surgical resection of incidental MD [18–20], the association with NETs remains rare, leading to inconclusive findings in reported studies in adults. Existing publications are lacking evidence-based guidelines, being absent particularly in pediatric cases.
Based on our experience, we recommend performing exploratory laparoscopy after the third recurrence of intussusceptions in children above 2 years old. Excision of the diverticulum upon identification is recommended to facilitate early recognition and management of carcinoid tumors, thereby preventing potential sequelae, or presenting with advanced stages.
MD remains a clinically significant entity, often associated with rare but potentially malignant complications such as NET. Timely recognition and surgical intervention are fundamental for optimal management and prevention of adverse outcomes in affected individuals.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Park JJ, Wolff BG, Tollefson MK, et al. Meckel diverticulum: the Mayo Clinic experience with 1476 patients (1950-2002). Ann Surg 2005;241:529–33. 10.1097/01.sla.0000154270.14308.5f.15729078 PMC 1356994 · doi ↗ · pubmed ↗
- 2An J, Zabbo CP. Meckel Diverticulum. In: Stat Pearls [Internet]. Treasure Island (FL): Stat Pearls Publishing. Available from: https://www.ncbi.nlm.nih.gov/books/NBK 499960/.29763135 · pubmed ↗
- 3Caracappa D, Gullà N, Lombardo F, et al. Incidental finding of carcinoid tumor on Meckel's diverticulum: case report and literature review, should prophylactic resection be recommended? World J Surg Oncol 2014;12:144. 10.1186/1477-7819-12-144.24884768 PMC 4031372 · doi ↗ · pubmed ↗
- 4Chaaban Y, Alhalabi R, Ba'Ath ME. A case report of axial torsion of Meckel's diverticulum: an acute abdomen without mechanical bowel obstruction in a child. Cureus 2024;16:e 69235. 10.7759/cureus.69235.39398822 PMC 11470226 · doi ↗ · pubmed ↗
- 5Chan CK, Pham T, Bhagat YV, et al. Incidental Meckel's diverticulum with neuroendocrine tumor. Cureus 2022;14:e 27625. 10.7759/cureus.27625.36072199 PMC 9437379 · doi ↗ · pubmed ↗
- 6De Andrade JP, Blakely AM, Nguyen AH, et al. Neuroendocrine Tumors of Meckel's diverticula: rare but fare well. Am Surg 2019;85:1125–8. 10.1177/000313481908501010.31657307 · doi ↗ · pubmed ↗
- 7Cingam SR, Kashyap S, Karanchi H. Carcinoid tumors. [Updated 2022 Sep 26]. In: Stat Pearls [Internet]. Treasure Island (FL): Stat Pearls Publishing; 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK 448101/.
- 8Modlin IM, Shapiro MD, Kidd M. An analysis of rare carcinoid tumors: clarifying these clinical conundrums. World J Surg 2005;29:92–101. 10.1007/s 00268-004-7443-z.15599742 · doi ↗ · pubmed ↗