Post-transcriptional regulation of aromatic amino acid metabolism by GcvB small RNA in Escherichia coli
Takeshi Kanda, Toshiko Sekijima, Masatoshi Miyakoshi

TL;DR
This paper explores how GcvB small RNA regulates aromatic amino acid metabolism in E. coli, identifying new target genes and mechanisms.
Contribution
The study identifies new GcvB target genes in AAA biosynthesis and transport and reveals a direct base pairing mechanism for aroG repression.
Findings
GcvB represses new target genes in AAA biosynthesis and transport via the R1 seed region.
GcvB directly inhibits aroG expression through base pairing with the R3 seed sequence.
GcvB overexpression affects growth and antibiotic resistance by regulating AAA transporters.
Abstract
Escherichia coli synthesizes aromatic amino acids (AAAs) through the common pathway to produce the precursor, chorismate, and the three terminal pathways to convert chorismate into Phe, Tyr, and Trp. E. coli also imports exogenous AAAs through five transporters. GcvB small RNA post-transcriptionally regulates more than 50 genes involved in amino acid uptake and biosynthesis in E. coli, but the full extent of GcvB regulon is still underestimated. This study examined all genes involved in AAA biosynthesis and transport using translation reporter assay and qRT-PCR analysis. In addition to previously verified targets, aroC, aroP, and trpE, we identified new target genes that were significantly repressed by GcvB primarily via the R1 seed region. Exceptionally, GcvB strongly inhibits the expression of aroG, which encodes the major isozyme of the first reaction in the common pathway, through…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
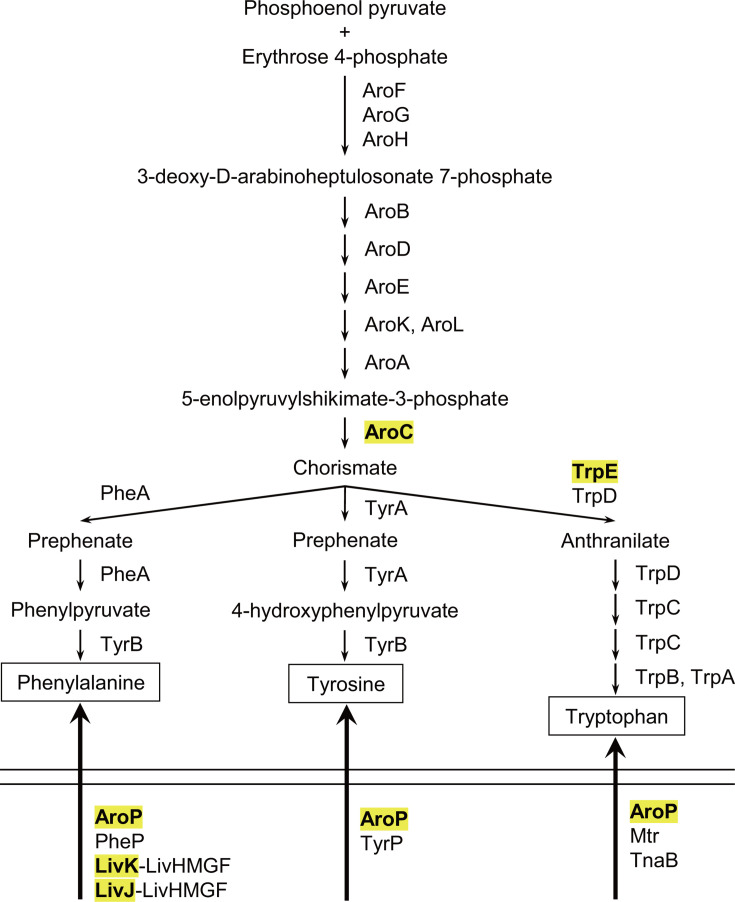 Fig 1
Fig 1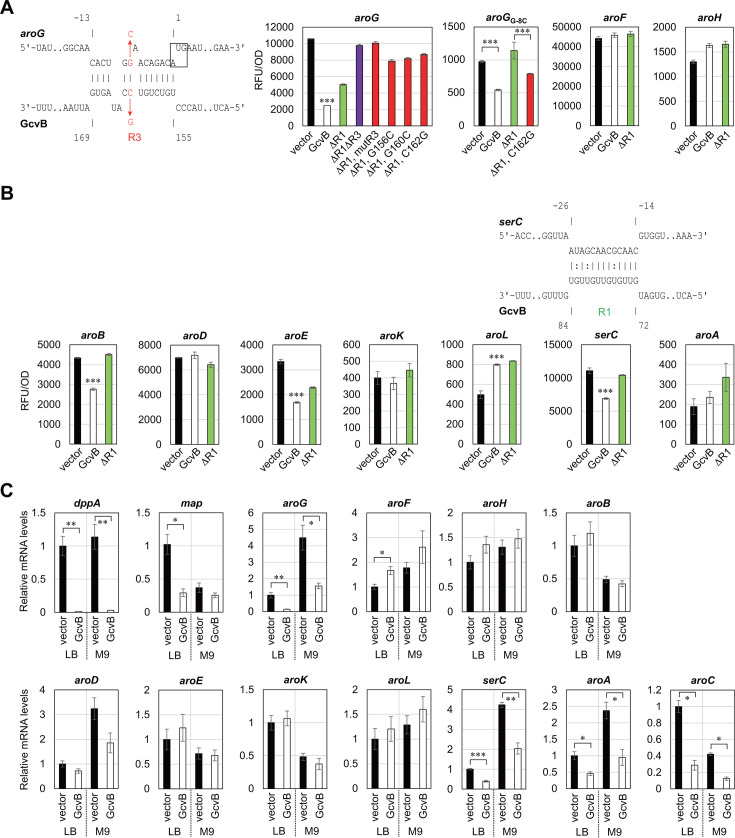 Fig 2
Fig 2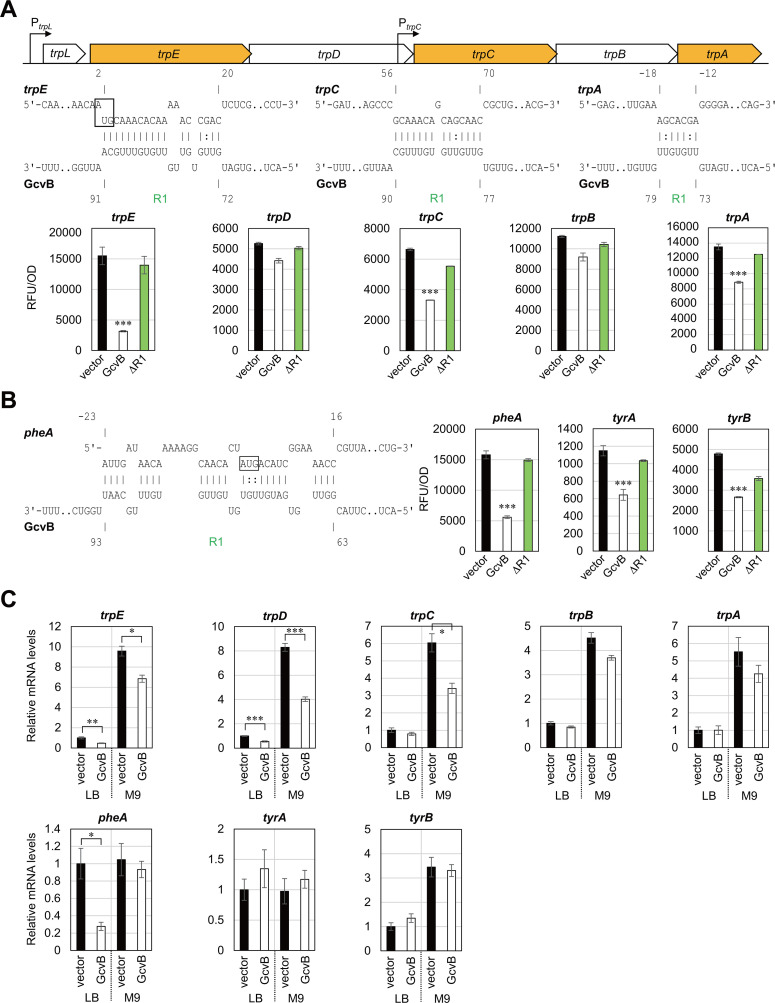 Fig 3
Fig 3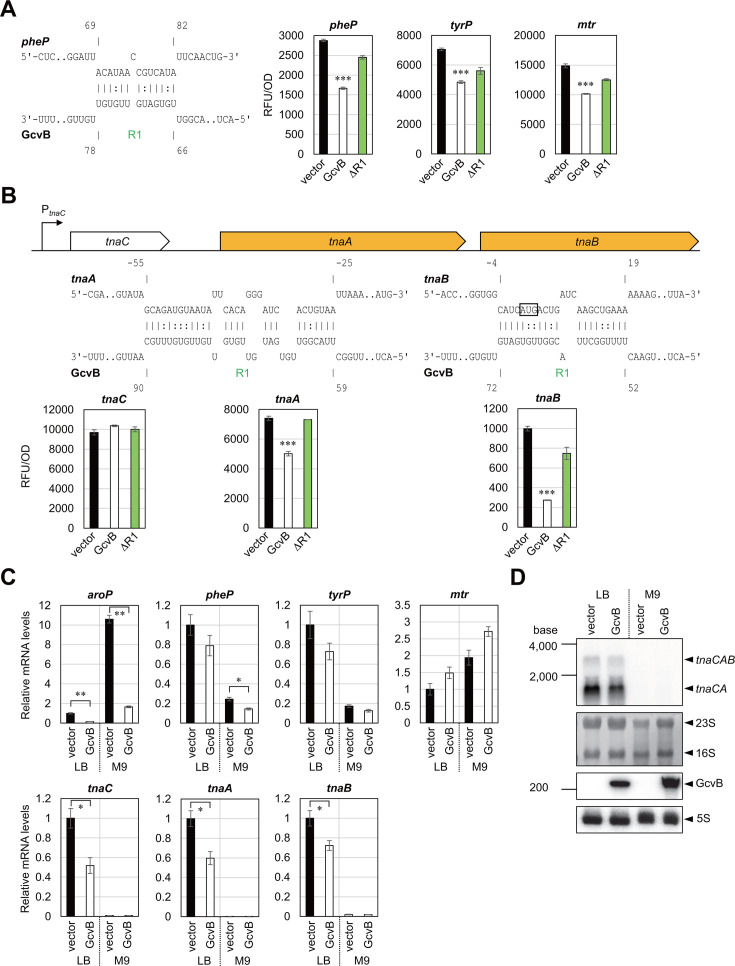 Fig 4
Fig 4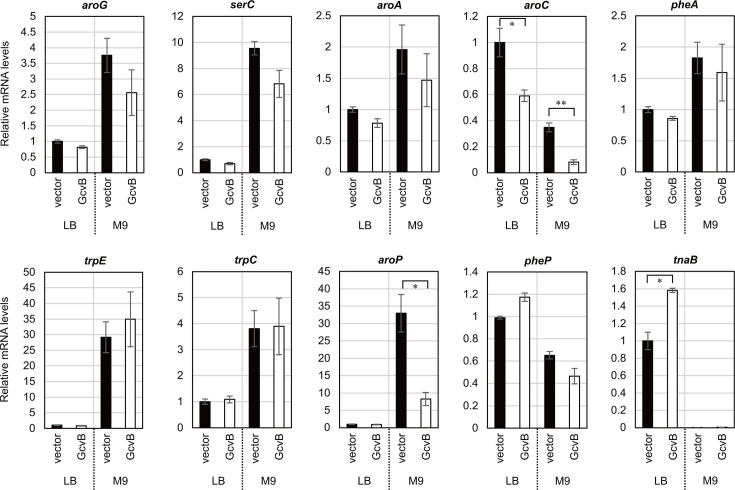 Fig 5
Fig 5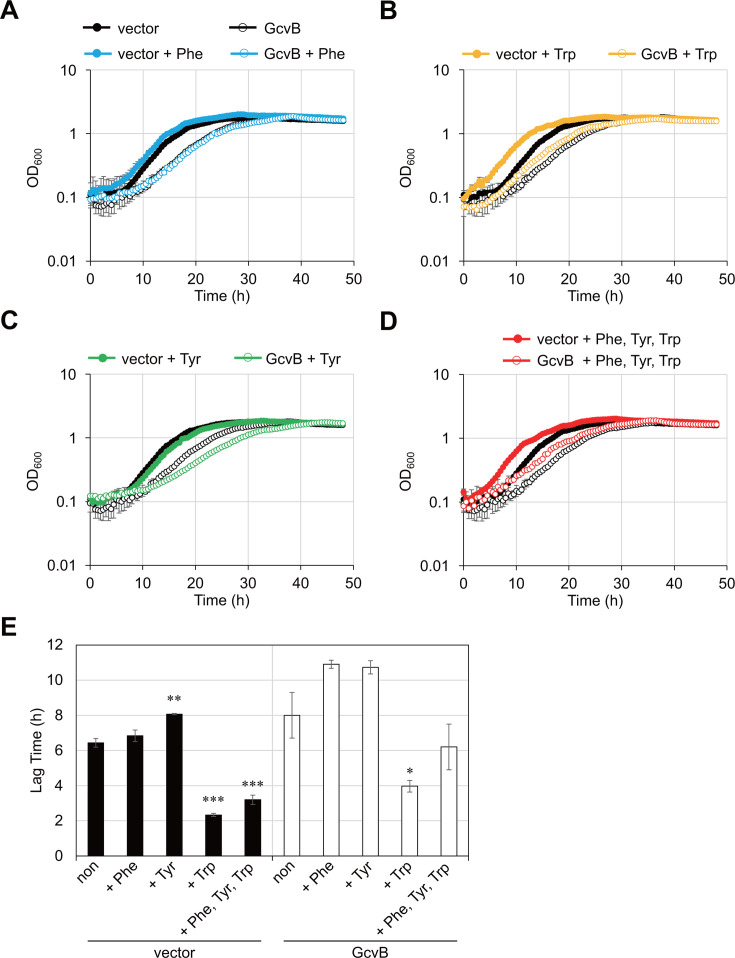 Fig 6
Fig 6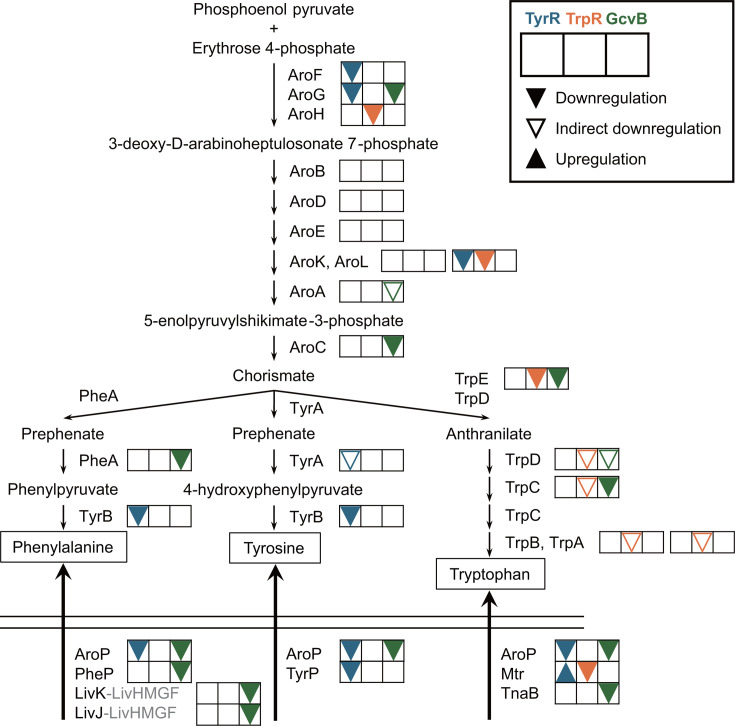 Fig 7
Fig 7| Gene | Reporter assay | qRT-PCR % mRNA ( | qRT-PCR % mRNA ( | IntaRNA | |||
|---|---|---|---|---|---|---|---|
| % RFU/OD (GcvB/vector) | LB | M9 | LB | M9 | Hybridization energy (kcal/mol) | GcvB | |
|
| 104 | 167 | 145 | −28.27 | 35–75 | ||
|
| 19.1 | 14.3 | 34.8 | 81.6 | 68.2 | −14.53 | 155–169 |
|
| 144 | 136 | 118 | −6.44 | 76–82 | ||
|
| 63.9 | 119 | 86.3 | −10.57 | 183–193 | ||
|
| 103 | 72 | 57.1 | −19.39 | 155–169 | ||
|
| 50.9 | 123 | 94.7 | −10.87 | 148–158 | ||
|
| 91.8 | 106 | 77.4 | −11.44 | 69–79 | ||
|
| 161 | 121 | 125 | −18.78 | 72–85 | ||
|
| 62.3 | 40.1 | 48.2 | 69.4 | 71.5 | −17.42 | 72–84 |
|
| 124 | 45.7 | 39.9 | 78.3 | 75 | −14.31 | 63–73 |
|
| 47.8 | 28.7 | 29.5 | 59 | 23.5 | −21.97 | 76–90 |
|
| 35.4 | 27.8 | 89 | 85.9 | 87.1 | −18.97 | 63–93 |
|
| 56 | 135 | 120 | −9.12 | 154–164 | ||
|
| 55.6 | 135 | 95.8 | −24.61 | 31-64 | ||
|
| 20.3 | 46.5 | 71.5 | 86.9 | 120 | −20.28 | 72–91 |
|
| 84 | 54.5 | 48.5 | −17.88 | 54–69 | ||
|
| 49.9 | 78.8 | 56.5 | 109 | 102 | −16.81 | 77–90 |
|
| 82 | 84.7 | 81.9 | −17.22 | 63–82 | ||
|
| 65.8 | 100 | 77 | −5.6 | 72–78 | ||
|
| 13.6 | 15.1 | 15.7 | 93.6 | 25.1 | −17.36 | 71–91 |
|
| 58.1 | 79 | 59.1 | 119 | 71.4 | −11.34 | 66–78 |
|
| 69 | 72.9 | 73.7 | −11.74 | 157–163 | ||
|
| 68.3 | 150 | 140 | −25.66 | 42–84 | ||
|
| 107 | 51.7 | 91.6 | −6.45 | 163–169 | ||
|
| 67.8 | 59.7 | 104 | −19.95 | 59–90 | ||
|
| 27.5 | 72.2 | 87 | 158 | 161 | −20.8 | 52–72 |
| Strain | Relevant markers/ genotype | Reference/ source |
|---|---|---|
| BW25113 | F-λ- | NBRP strain |
| Δ | BW25113 Δ | ( |
| ΤΜ529 | W3110 | ( |
| Δ | Δ | This study |
- —MEXT | Japan Society for the Promotion of Science (JSPS)
- —MEXT | Japan Society for the Promotion of Science (JSPS)
- —MEXT | Japan Society for the Promotion of Science (JSPS)
- —Institute for Fermentation, Osaka (IFO)
- —Kato Memorial Bioscience Foundation
- —Mishima Kaiun Memorial Foundation
- —Asahi Group | Asahi Group Foundation (Asahi Group Research Foundation)
- —Takeda Science Foundation (TSF)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Metabolic Engineering and Bioproduction · Bacterial Genetics and Biotechnology · RNA and protein synthesis mechanisms
INTRODUCTION
Aromatic amino acids (AAAs), namely l-phenylalanine (Phe), l-tyrosine (Tyr), and l-tryptophan (Trp), are important for food, pharmaceutical, and chemical industries. Escherichia coli is genetically engineered in its biosynthesis and transport pathways and is widely used for fermentative production of AAAs (1–4). The biosynthetic pathway of AAAs is composed of the common pathway and three terminal pathways (5). The common pathway, a.k.a. the shikimate pathway, starts from the condensation of phosphoenolpyruvate and erythrose 4-phosphate, which is catalyzed by 3-deoxy-d-arabino-heptulosonate 7-phosphate (DAHP) synthase isozymes, AroG, AroF, and AroH (Fig. 1). DAHP is then converted to chorismate, the common precursor of the three AAAs, through six enzymatic reactions. Chorismate is transformed to phenylpyruvate and 4-hydroxyphenylpyruvate by chorismate mutase-prephenate dehydratase (PheA) and chorismate mutase-prephenate dehydrogenase (TyrA) and subsequently to Phe and Tyr by aromatic aminotransferase (TyrB), respectively. Trp is produced from chorismate via anthranilate by the enzymes encoded in the trpEDCBA operon. Since the biosynthesis of AAAs requires chemical intermediates from glycolytic pathway and thus is costly (6), E. coli imports exogenous AAAs through five different transport systems when available (Fig. 1). The general permease AroP transports all three AAAs with high affinities, but E. coli also possesses four dedicated transporters: PheP for Phe, TyrP for Tyr, and Mtr and TnaB for Trp (5).
Schematic representation of AAA biosynthesis and import pathway in E. coli. The proteins whose expression is directly regulated by GcvB are highlighted in yellow.
Fundamental study of the regulatory mechanism underlying the metabolic pathways is crucial to improving E. coli’s efficiency and yields for the fermentative production of valuable compounds. The main control of AAA biosynthetic pathways is feedback inhibition of the first reaction in each pathway by the end products (5). Three isozymes in the common pathway, AroG, AroF, and AroH, are allosterically inhibited by Phe, Tyr, and Trp, respectively. PheA, TyrA, and TrpE of the branched pathways are also allosterically inhibited by the respective end products. Next, the gene expression of AAA biosynthetic enzymes and transporters is regulated mainly at the level of transcription. Two transcriptional regulatory proteins, TyrR and TrpR, control the transcription of genes dispersed at different loci in the E. coli genome (5, 7). In addition, the pheA and trpE genes are preceded by transcriptional attenuators, in which the leader mRNAs contain consecutive Phe and Trp codons (8).
Transcriptional and post-transcriptional regulation act together to fine-tune the expression of amino acid metabolic genes (9). The global regulatory small RNA (sRNA) GcvB is transcriptionally regulated by the glycine cleavage pathway regulators, GcvA and GcvR (10), and is one of the most abundantly expressed sRNAs bound with the RNA chaperone Hfq in nutrient-rich media. GcvB contains three seed sequences, R1, R2, and R3, to hybridize with its target mRNAs in trans and represses their expression mainly at the level of translation initiation (11–19). By compiling the global RNA–RNA interactome data sets (20–23), we have estimated that GcvB interacts with >50 mRNAs in E. coli, the majority of which are associated with transport and biosynthesis of many but not all amino acids (24). However, amino acid biosynthetic genes are underrepresented in the data sets due to their low expression levels during growth in nutrient-rich media.
Regarding AAA biosynthesis and transport, we have verified that GcvB inhibits the expression of aroC, aroP, and trpE at the post-transcriptional level (24). However, it is possible that GcvB redundantly regulates more genes in the same pathway. This study aimed to determine the full extent of GcvB targets involved in the AAA metabolism in E. coli. To this end, we constructed translational reporters for all AAA biosynthetic and transporter genes and validated additional genes that are post-transcriptionally inhibited by GcvB via either R1 or R3 seed sequence.
RESULTS
Regulation of the common biosynthetic pathway
We have previously shown that GcvB post-transcriptionally represses the expression of AroC (24), which catalyzes the last step in the common biosynthetic pathway to produce chorismate, the precursor of AAAs. To investigate the post-transcriptional regulation of the other enzymes in the common pathway, we applied the established translational GFP reporter system (25, 26). The translational fusions with the superfolder GFP (sfGFP) were constructed based on pXG-10sf and pXG-30sf plasmids (Tables S1 through S4). Here, we used E. coli BW25113 ΔgcvBΔsroC mutant as the host strain for the reporter plasmids and the compatible GcvB-expressing plasmids because the SroC sRNA induces the RNase E-mediated degradation of GcvB by binding at two distant regions (27) and may alter the steady-state levels of GcvB variants in this assay (24).
In E. coli, the first reaction is catalyzed by the three DAHP synthase isozymes, AroG, AroF, and AroH, among which AroG contributes approximately 80% of DAHP production (28). We found that the expression level of aroG was reduced to ~20% upon GcvB expression, while aroF and aroH were not significantly regulated (Fig. 2A). The translation initiation region of aroG was predicted to base pair with the R3 seed region of GcvB by the IntaRNA program (29). To verify the interaction between aroG and R3, we took advantage of the previously constructed plasmids expressing various GcvB mutants (24). The repression of aroG was slightly relieved by deletion of the major R1 seed region (ΔR1) and was fully abrogated by further deletion of R3 (ΔR1ΔR3) (Fig. 2A). Moreover, a five-nucleotide replacement (mutR3) and single-nucleotide substitutions (G156C, G160C, and C162G) in the R3 seed sequence disrupted the repression by GcvBΔR1 as expected. Finally, the G-8C mutation in the Shine–Dalgarno sequence of aroG (aroGG-8C), which reduced the translation efficiency by ~10-fold, disabled the repression by GcvBΔR1, but the complementary mutation C162G in GcvBΔR1 partially restored the repression of aroGG-8C (Fig. 2A). These results indicate that the R3 seed region of GcvB directly interacts with the translation initiation region of aroG to inhibit its expression.
*Post-transcriptional regulation of the common biosynthetic pathway mediated by GcvB. (A, B) GFP reporter assays in E. coli ΔgcvBΔsroC strain harboring pTP11 (vector), pPL-gcvB (GcvB) or its derivatives. ΔR1: deletion mutant of the R1 seed region. ΔR1ΔR3: a combination of deletions of the R1 and R3 seed regions. mutR3, G156C, G160C, and C162G: nucleotide substitution mutations in the R3 seed region. GFP fluorescence of overnight-grown cells in LB medium was measured and divided by OD600 for normalization. Base-pairing interaction between aroG mRNA (upper) and GcvB (below) was predicted by the IntaRNA program. Numbers above and below the nucleotide sequences indicate the nt location relative to the start codon of the mRNA and the transcription start site of GcvB, respectively. The start codon of aroG is shown in a box. (C) Relative mRNA levels quantified by qRT-PCR. E. coli ΔgcvBΔsroC strain harboring pKP8-35 (vector) or pBAD-gcvB (GcvB) was grown in LB medium or M9 minimal medium supplemented with 0.2% glycerol. At exponential phase (OD660 = 0.5), 0.2% l-arabinose was added to the cultures to induce GcvB expression, and after 10 min, total RNA was isolated. Values are presented as mean ± standard error from three independent experiments (n = 3) and were statistically analyzed using one-way ANOVA with Bonferroni post hoc test in (A, B), or using the two-tailed Student’s t-test in (C) (*P < 0.05, **P < 0.01, **P < 0.001).
The remaining six genes of the common pathway were also examined by the two-plasmid reporter assay. GcvB moderately repressed aroB (64%) and aroE (51%), but aroD, aroK, and aroA were not significantly affected (Fig. 2B). Deletion of R1 slightly relieved the repression of aroB and aroE, but we could not find firm base-pairing interactions within the R1 seed region (Table 1). The translational fusion of aroL was activated by ~1.6-fold in an R1-independent manner for an unknown reason.
To further confirm whether the levels of target mRNAs are affected by GcvB, we performed qRT-PCR analysis. The E. coli ΔgcvBΔsroC strain was transformed by pBAD-gcvB and was grown in LB medium or M9 minimal medium supplemented with 0.2% glycerol until the optical density (OD) reached 0.5, and then the expression of GcvB was induced by adding 0.2% l-arabinose for 10 min. We confirmed that the known GcvB targets, dppA and map, were significantly downregulated by the pulse expression of GcvB (Fig. 2C). It is noteworthy that the map mRNA level was dropped to ~30% despite only slight repression by GcvB at the translational level (24). Among the common pathway genes, only aroG, aroC, and aroA were significantly repressed by GcvB (Fig. 2C). Since aroA is cotranscribed with the upstream serC gene encoding the phosphoserine/phosphohydroxythreonine aminotransferase for serine and pyridoxal 5′-phosphate biosynthesis (30–32), GcvB is likely to regulate serC as the direct target. The 5′UTR of serC was predicted to interact strongly with the R1 seed of GcvB, and the translational fusion of serC was significantly repressed by GcvB in an R1-dependent manner (Fig. 2B). The levels of serC mRNA were affected by GcvB similarly to aroA (Fig. 2C). These results indicate that GcvB induces the degradation of serC-aroA bicistronic mRNA by directly targeting serC.
Regulation of terminal biosynthetic pathways
The enzymes of Trp biosynthetic pathway are encoded by the trp operon composed of the trpEDCBA structural genes preceded by the trpL attenuator (33). The trp operon is transcribed mainly from the trpL promoter under the control of TrpR repressor, and the downstream genes are additionally transcribed from the internal trpC promoter (34, 35). We have previously found that GcvB represses the first structural gene trpE via the R1 seed region (24). To examine whether GcvB directly regulates the expression of downstream genes, we constructed the translational fusions of intraoperonic regions using the pXG-30sf vector (26). Ectopic expression of GcvB significantly downregulated trpC and trpA in an R1-dependent manner (Fig. 3A). The IntaRNA program predicted the base pairing between the R1 seed region of GcvB and the coding region of trpC. However, according to the IntaRNA program, the interaction between the GcvB R1 seed and the trpB-A intergenic region cloned in the translational reporter plasmid was only seven base pairs including two wobble base pairs (Fig. 3A). qRT-PCR analysis confirmed that the mRNA levels of trp genes were elevated in the absence of Trp, and pulse expression of GcvB significantly downregulated the upstream three genes of trp operon but not trpB and trpA (Fig. 3C). This result is in line with the previous reports that the promoter-proximal segments of trpEDCBA mRNA is degraded faster than the promoter-distal segments (36, 37). We suggest that GcvB post-transcriptionally regulates the trp operon mainly by binding two different sites in trpE and trpC via the R1 seed region.
*Post-transcriptional regulation of the terminal biosynthetic pathways mediated by GcvB. (A, B) GFP reporter assays in E. coli ΔgcvBΔsroC strain harboring pTP11 (vector), pPL-gcvB (GcvB), or pPL-gcvB ΔR1. GFP fluorescence of overnight-grown cells in LB medium was measured and divided by OD600 for normalization. A schematic of the trp operon is provided above in (A). Base-pairing interactions between target mRNAs (upper) and GcvB (below) were predicted by the IntaRNA program. Numbers above and below the nucleotide sequences indicate the nt location relative to the start codon of the mRNA and the transcription start site of GcvB, respectively. The start codons of trpE and pheA are shown in a box. (C) Relative mRNA levels quantified by qRT-PCR as in Fig. 2C. Values are presented as mean ± standard error from three independent experiments (n = 3) and were statistically analyzed using one-way ANOVA with Bonferroni post hoc test in (A, B), or using the two-tailed Student’s t-test in (C) (*P < 0.05, **P < 0.01, **P < 0.001).
Previously, the MS2-affinity purification coupled with RNA sequencing (MAPS) analysis has revealed that GcvB significantly interacts with pheA and tyrA (20), both of which encode the first enzymes for Phe and Tyr biosynthetic pathways, respectively (5). A recent RNA interaction by ligation and sequencing (RIL-seq) study in an enteropathogenic E. coli has also detected the interaction between pheA and GcvB (38). The tyrA gene is located downstream of aroF, and the aroF promoter is transcriptionally regulated by TyrR in response to Tyr as a corepressor (39, 40). The transamination reaction to synthesize Phe and Tyr is preferentially catalyzed by TyrB, whose transcription is also under the control of TyrR (41). To verify whether GcvB regulates the Phe and Tyr biosynthetic pathways, the translational fusions of pheA, tyrA, and tyrB were constructed and examined by the reporter analysis. pheA was significantly repressed by GcvB, and as predicted, the R1 seed region is involved in the repression (Fig. 3B). GcvB moderately repressed tyrA and tyrB in an R1-dependent manner, but we could not find any base-pairing interactions within the R1 seed region (Table 1). qRT-PCR analysis showed that the pulse expression of GcvB decreased the mRNA levels of pheA exclusively in LB medium but had no effect in M9 medium, indicating a condition-specific regulation. In contrast, the mRNA levels of aroF-tyrA and tyrB were not significantly affected upon GcvB expression ([Fig. 2C and 3C](#F2 F3)). Altogether, we suggest that GcvB represses the expression of first enzymes of the Trp and Phe terminal pathways but not that of Tyr (see the section “GcvB inhibits biosynthesis and import of AAAs”).
Regulation of AAA transporters
Among the five AAA transporters in E. coli, only aroP has previously been identified as the target of GcvB (24). Nonetheless, because GcvB redundantly regulates more than half of known amino acid transporters in E. coli (24), GcvB potentially interacts with mRNAs of the other AAA transporter genes. The R1 seed region is predicted to interact with tnaB, pheP, and mtr, but tyrP may only form a 7 bp hybrid with the R3 seed region (Table 1). The reporter analysis showed that GcvB strikingly repressed tnaB (28%) but only modestly pheP, tyrP, and mtr (Fig. 4A). qRT-PCR analysis verified significant downregulation of pheP and tnaB upon GcvB pulse expression during growth in M9 and LB media, respectively (Fig. 4C).
*Post-transcriptional regulation of the AAA transporters mediated by GcvB. (A, B) GFP reporter assays in E. coli ΔgcvBΔsroC strain harboring pTP11 (vector), pPL-gcvB (GcvB) or pPL-gcvB ΔR1. GFP fluorescence of overnight-grown cells in LB medium was measured and divided by OD600 for normalization. A schematic of the tna operon is provided above in (B). Base-pairing interactions between target mRNAs (upper) and GcvB (below) were predicted by the IntaRNA program. Numbers above and below the nucleotide sequences indicate the nt location relative to the start codon of the mRNA and the transcription start site of GcvB, respectively. The start codon of tnaB is shown in a box. (C) Relative mRNA levels quantified by qRT-PCR as in Fig. 2C. Values are presented as mean ± standard error from three independent experiments (n = 3) and were statistically analyzed using one-way ANOVA with Bonferroni post hoc test in (A, B), or using the two-tailed Student’s t-test in (C) (*P < 0.05, **P < 0.01, **P < 0.001). (D) GcvB induces degradation of the tnaCAB mRNA. The same RNA samples as in (C) were analyzed by northern blotting. In each lane, 5 µg of total RNA was loaded and hybridized with 32P-labeled antisense RNA probe targeting tnaA mRNA, GcvB sRNA, and 5S rRNA. 23S and 16S rRNA stained with methylene blue on the blot are indicated as loading controls.
The polycistronic tnaCAB mRNA encodes the leader peptide, tryptophanase, responsible for the degradation of Trp into indole, and the Trp:H^+^ symporter, respectively (42). It has been shown by the cross-linking, ligation, and sequencing of hybrids (CLASH) analysis that the tnaCAB mRNA interacts with GcvB at multiple sites (21). GcvB moderately repressed tnaA (68%), and deletion of R1 abrogated the repression (Fig. 4B). This result is in accordance with the R1-mediated interaction as predicted by IntaRNA. qRT-PCR analysis revealed that tnaCAB was highly expressed during growth in LB medium, and the mRNA levels at three coding regions of tnaC, tnaA, and tnaB were reduced by approximately twofold upon GcvB pulse expression (Fig. 4C). Northern blotting with a specific probe targeting tnaA mRNA identified two forms of the transcripts, tnaCAB (~3.1 kb) and tnaCA (~1.8 kb), and both transcripts were reduced by approximately twofold upon GcvB pulse expression (Fig. 4D). These results suggest that GcvB induces the degradation of overall tna operon mRNA levels.
RNase E is responsible for the degradation of GcvB target mRNAs
It has been empirically shown that Hfq-dependent sRNAs exert post-transcriptional regulation primarily at the level of translation initiation, and in many cases, translational regulation accompanies secondary effects on the mRNA stability. Previous studies have shown that GcvB actively induces RNase E-mediated degradation of many target mRNAs (18, 20, 43).
The above results revealed a total of nine direct GcvB targets involved in AAA biosynthesis and transport, aroG, aroC, serC, trpE, trpC, pheA, aroP, pheP, and tnaB, while GcvB is likely to repress aroA indirectly. To verify whether RNase E is involved in the degradation of the target mRNAs upon GcvB pulse expression, we performed qRT-PCR analysis in the rne598 background devoid of the C-terminal domain of RNase E (44), which interacts with Hfq and serves as a scaffold for sRNA-mediated target mRNA decay (45). Since GcvB itself is degraded by RNase E through interaction with SroC (27), we analyzed the target mRNA levels in the ΔsroC genetic background. As expected, the verified target mRNAs except aroC and aroP became insensitive to GcvB pulse expression (Fig. 5). This result indicates that RNase E is required for the decay of many GcvB targets but also implicates the other RNases in the post-transcriptional regulation of aroC and aroP.
*Degradation of most target mRNAs of GcvB is dependent on RNase E. Relative mRNA levels quantified by qRT-PCR as in Fig. 2C, except that the rne598 mutation was introduced as the background. Values are presented as mean ± standard error from three independent experiments (n = 3) and were statistically analyzed using the two-tailed Student’s t-test (*P < 0.05, *P < 0.01).
GcvB inhibits biosynthesis and import of AAAs
Since GcvB represses the expression of several AAA biosynthetic enzymes and transporters, we hypothesized that overexpression of GcvB results in growth defects in a minimal medium. To test this, we compared the growth of E. coli ΔgcvBΔsroC strains harboring pBAD-gcvB or its vector control in M9 minimal medium supplemented with each AAA. The addition of Phe or Trp improved the growth of the control strain (Fig. 6A and B). By contrast, Tyr posed a growth delay (Fig. 6C), which was completely restored by the addition of Phe and Trp (Fig. 6D). This result suggests that Tyr inhibits the other AAA synthetic enzymes.
*GcvB induces growth defect by repressing import of AAAs under amino acid-limiting conditions. E. coli ΔgcvBΔsroC strain harboring pKP8-35 (vector) or pBAD-gcvB (GcvB) was grown in M9 minimal medium supplemented with 0.2% glycerol and 0.2% l-arabinose in the presence of (A) Phe, (B) Trp, (C) Tyr, and (D) a combination of the three AAAs (1 mM). Growth was continuously monitored by measuring OD600 every 30 min for 48 h. OD600 values of the vector control and the GcvB-expressing strain in the absence of AAAs are shown as black closed circles and black open circles, respectively, as controls in (A–D). The calculated lag times based on these data are shown in (E). Data are presented as mean ± standard error from three independent experiments (n = 3) and were statistically analyzed using the one-way ANOVA with Bonferroni post hoc test (*P < 0.05, **P < 0.01, **P < 0.001).
The ectopic expression of GcvB generally extended the lag phase in the presence of AAAs (Fig. 6E), suggesting that GcvB inhibits the import of AAAs. Moreover, Tyr caused a more severe growth inhibition on the GcvB-expressing strain (Fig. 6C). The growth rate during exponential phase (h^−1^) of the GcvB-expressing strain was 0.24 in the absence of Tyr and was decreased to 0.16 in its presence, although Tyr had no significant effect on the growth rate of the vector control (absence of Tyr: 0.33, presence of Tyr: 0.31). This result suggests that, in addition to the Tyr-driven feedback inhibition, GcvB inhibits the expression of AAA synthetic pathways other than the Tyr terminal pathway.
Azaserine (O-diazoacetyl-l-serine), an antibiotic compound produced by Streptomyces fragilis, is taken up through AroP and the ABC transporter LivJKHMGF to inhibit the growth of E. coli (46, 47). To test whether GcvB alleviates the toxicity of azaserine by limiting its import, we determined the minimal inhibitory concentration (MIC) of E. coli ΔgcvBΔsroC strains harboring pP_L_-gcvB or its vector control in M9 minimal medium containing 0.4% glucose. The MIC of azaserine was 30 µM in the strain harboring the control vector, and the GcvB-expressing strain exhibited a twofold higher MIC of 60 µM. Altogether, these results indicate that GcvB inhibits the uptake of AAAs.
DISCUSSION
This study shows that GcvB post-transcriptionally regulates AAA metabolism at several key steps (Fig. 7). Together with our previous report (24), we conclude that GcvB represses a total of six biosynthetic genes (aroG, aroC, serC-aroA, trpE, trpC, and pheA) and three transporter genes (aroP, pheP, and tnaB) as summarized in Table 1. Many of these target genes are downregulated via the conserved R1 seed region of GcvB. Nonetheless, GcvB interacts with the translation initiation region of aroG mRNA with the R3 seed region to repress the expression of the first enzyme of the common pathway. For aroB, aroE, aroL, trpA, tyrA, tyrB, tyrP, and mtr, the expression levels of translational fusions were affected by GcvB overexpression, but their mRNA levels were not significantly downregulated (Table 1). It is possible that these genes are regulated by GcvB mainly at the translational level, but the predicted base-pairing regions are not located near the R1 seed region. GcvB might possess sequences other than the known three conserved seeds to interact with the target mRNAs.
Schematic representation of the transcriptional and post-transcriptional regulation of the AAA biosynthesis and import pathways in E. coli. Transcriptional regulation by TyrR and TrpR and post-transcriptional regulation by GcvB are shown next to the corresponding enzymes. ▼: direct downregulation, ▽: indirect downregulation via upstream gene regulation, ▲: direct upregulation.
We observe a substantial overlap between the transcriptional and post-transcriptional regulons of amino acid metabolism in E. coli (9). Both TyrR and GcvB repress aroG and aroP, which encode the major DAHP synthase and the major AAA transporter, respectively, while TrpR and GcvB together repress the trp operon (Fig. 7). Interestingly, the Trp and Phe biosynthetic genes are regulated at the post-transcriptional level both by the small RNA and the transcriptional attenuators, while the Tyr biosynthetic genes are predominantly regulated at the transcriptional level. In addition, the global transcriptional regulator Lrp regulates one-third of the E. coli genome, but aroG is the only direct target of Lrp among the AAA metabolic genes (48). Lrp activates the transcription of aroG in the absence of leucine and dissociates from the promoter in the presence of leucine (49). We demonstrate that the translation of aroG is directly repressed by GcvB via the R3 seed sequence (Fig. 2A). To control the biosynthesis of AAAs, the first reaction of the common pathway is critical. Since AroG is the major isozyme and is allosterically inhibited by Phe, a gcvB mutation combined with the feedback resistance mutation in aroG (50) will be beneficial for the fermentative production of AAAs. By contrast, the last reaction of the common pathway is also rate-limiting. However, the expression of aroC is not regulated by any transcriptional regulators and has long been believed to be constitutive in E. coli (5). We have previously reported that GcvB downregulates the expression of aroC in the prmB-aroC-mepA-yfcA-epmC-yfcL gene cluster, leaving the expression level of prmB constant (24). Therefore, we conclude that GcvB is the crucial sRNA regulator of AAA biosynthesis by targeting the first and last steps in the common pathway.
Post-transcriptional regulation of AAA biosynthetic pathways extends through two additional sRNAs. An Hfq-dependent sRNA RydC primarily activates the expression of cyclopropane fatty acid synthase by stabilizing the cfa mRNA (51) and modestly regulates trpE and pheA using different seed sequences (52). The expression of RydC is regulated by a GntR family transcriptional repressor YieP, whose signaling molecule is unknown (53). An Hfq-independent sRNA RybA is induced by peroxide stress and represses aroF and aroL in a TyrR-dependent manner without affecting the level of tyrR mRNA (54). RybA encodes a small peptide MntS, which is involved in manganese homeostasis by inhibiting the MntP transporter (55, 56). Further study is required to understand how significantly these sRNAs along with GcvB regulate AAA metabolism and correlate it with the other biological pathways.
Identifying the direct targets of sRNAs is important to understand the impact of post-transcriptional regulation through the base-pairing mechanism. GcvB is widely conserved in Enterobacteriaceae and the other families of γ-Proteobacteria and has been regarded as one of the model sRNAs targeting multiple mRNAs. Pioneering multi-omics studies in Salmonella revealed that GcvB specifically represses the substrate-binding components of amino acid ABC transporters among the proteome of periplasmic fraction and alters the transcriptome as translation inhibition induces degradation of most target mRNAs (11, 12). In addition, a bioinformatic search allowed to discover many interactions with the G/U-rich R1 seed sequence of GcvB (12). Recently, base-pairing interactions between RNAs can be experimentally identified through advanced transcriptomic techniques (57, 58). We have previously compiled the RNA–RNA interactome data sets to identify new GcvB targets, raising the number of validated GcvB targets to more than 50 (24). However, as aroG was predicted as a target through a bioinformatic approach (59) but did not fit the criteria adopted in our previous survey (24), genuine GcvB targets are still underrepresented in the previous RNA–RNA interactome data sets due to low expression levels. This study reveals new sRNA targets by thoroughly examining genes of particular metabolic functions and thus highlights the importance of laborious reporter assays dedicated to sRNA-mediated post-transcriptional regulation.
The criteria to discriminate between targets and non-targets of sRNAs can be somewhat arbitrary. The previously identified GcvB target, map, is translationally repressed to ~70% via the R3 seed region (24), but its mRNA level is strikingly downregulated to 30% (Fig. 2C). By contrast, the mRNA levels of aroB, aroE, trpA, tyrA, and tyrB were not significantly altered upon GcvB pulse expression, but their translational fusions were substantially repressed (Table 1), suggesting that these genes might be regulated solely at the translational level. This type of sRNA targets cannot be identified merely by transcriptomics but by proteomics or ribosome profiling (Ribo-seq) (60), as represented by the translational regulation of lpp by MicL sRNA (61). Ribo-seq analysis has revealed that RyhB sRNA regulates >50 genes involved in iron utilization but does not always alter the level of target mRNAs (62). Interestingly, although GcvB abundantly interacts with the raiA mRNA encoding the ribosome-associated inhibitor (20–24), Faigenbaum-Romm et al. have shown that GcvB overexpression exerts no regulatory output on the raiA mRNA, arguing that a subset of the RNA–RNA interactions are sporadic (63). To complete the identification of sRNA regulon, we require more RNA–RNA interactome data sets and expression profiles at both transcription and translation levels under various growth conditions relevant to the sRNA of interest and further validation in physiological significance using standard methods.
MATERIALS AND METHODS
Bacterial strains and growth media
The strains used in this study are listed in Table 2. Bacterial cells were grown at 37°C with reciprocal shaking at 180 rpm in LB Miller medium (Nacalai Tesque) or M9 minimal medium supplemented with 0.2% glycerol. Where required, media were supplemented with antibiotics at the following concentrations: 50 µg/mL ampicillin, 50 µg/mL kanamycin, and 12.5 µg/mL chloramphenicol. To construct the ΔgcvBΔsroC rne598 strain, the rne598-FLAG-cat allele was transduced using P1 phage from strain TM529 (44) into the ΔgcvBΔsroC strain. The cat cassette was removed by introducing pCP20, followed by curation during growth at 37°C.
Oligonucleotides and plasmids
The plasmids and oligonucleotides used in this study are listed in Tables S1 and S2, respectively. The plasmids for constitutive expression of GcvB (pP_L_-gcvB) or its derivatives were constructed previously (24). The l-arabinose-inducible expression plasmid for E. coli GcvB (pBAD-gcvB) was constructed using the primers JVO-0895 and MMO-0086 as previously described (12). Translational fusions were constructed as previously described (25, 26).
GFP fluorescence quantification
The E. coli BW25113 ΔgcvBΔsroC strains harboring a combination of the sfgfp translational fusions and pP_L_-gcvB or its derivatives were inoculated from single colonies into 400 µL LB medium in 96 deep-well plates (Thermo Scientific) and were grown overnight at 37℃ with rotary shaking at 1,200 rpm in DWMax M-BR-032P plate shaker (Taitec). A 100 µL aliquot of the overnight cultures was transferred into 96-well optical bottom black microtiter plates (Thermo Scientific), and both optical density at 600 nm (OD_600_) and fluorescence (excitation at 485 nm and emission at 535 nm with dichroic mirror of 510 nm, fixed gain value of 50) were measured using Spark plate reader (Tecan). The relative fluorescence unit (RFU) was calculated by subtracting the autofluorescence of bacterial cells of the same strains harboring the GcvB-expressing plasmids.
Total RNA extraction
The E. coli BW25113 ΔgcvBΔsroC strains harboring pBAD-gcvB or its vector control (pKP8-35) (64) were grown in LB medium or M9 minimal medium supplemented with 0.2% glycerol. When the OD_660_ reached 0.5, l-arabinose was added to the cultures at a final concentration of 0.2% to induce GcvB expression. After 10 min, two volumes of RNA Protect Bacterial Reagent (Qiagen) was added to one volume of the cultures to stabilize cellular RNA. Total RNA was isolated using NucleoSpin RNA (Macherey–Nagel) according to the manufacturer’s instruction. The RNA samples were treated with DNase I at room temperature for 30 min.
qRT-PCR
cDNA was synthesized using ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo). qRT-PCR was performed using TB Green Premix Ex Taq™ II (Takara Bio) on QuantStudio 5 Real-Time PCR System (Thermo Scientific). Each target-gene mRNA level was normalized to a reference gene transcript (16S rRNA) from the same RNA sample. Fold changes were determined using the 2^−ΔΔ^^Ct^ method (65). The sequences of the primers used are shown in Table S2.
Northern blotting
For mRNA analysis, total RNA (5 µg) was separated by 1.5% agarose gel electrophoresis in the presence of formaldehyde. DynaMarker Prestain Marker for RNA high (BioDynamics Laboratory) was used as a size marker. The gels were transferred onto Nytran SuPerCharge nylon membranes (Cytiva) overnight by capillary blotting using TurboBlotter Kit (Cytiva). The membranes were crosslinked with transferred RNA by UV light at 120 mJ/cm^2^. Before probe hybridization, the membranes were stained with 0.3 M sodium acetate containing 0.03% methylene blue. A 161 bp [^32^P]-labeled antisense RNA probe targeting tnaA mRNA was synthesized by in vitro transcription using the MAXIscript kit (Invitrogen). The specific DNA fragments required for probe generation were amplified by PCR with the primers MMO-1062 and MMO-1704 (Table S2). Prehybridization (1 h) and hybridization (24 h) were performed at 70°C in ULTRAhyb ultrasensitive hybridization buffer (Thermo Scientific). The membranes were subsequently washed in 2 × SSC/0.1% SDS at 20°C for 20 min, 1 × SSC/0.1% SDS at 65°C for 15 min, and 0.5 × SSC/0.1% SDS at 65°C for 15 min.
For sRNA analysis, total RNA was isolated using the TRIzol reagent (Invitrogen) and precipitated with isopropanol and cold ethanol. RNA was quantified using NanoDrop One (Invitrogen). Total RNA (5 µg) was separated by 6% polyacrylamide/7 M urea gel electrophoresis in 1 × TBE buffer. DynaMarker RNA Low II ssRNA fragments (BioDynamics Laboratory) was used as a size marker. The gels were transferred onto Hybond-XL nylon membrane (GE Healthcare) by electroblotting. The membrane was crosslinked with transferred RNA by 120 mJ/cm^2^ UV light, incubated for prehybridization in Rapid-Hyb buffer (Amersham) at 42°C for 1 h, and then incubated for hybridization with a [^32^P]-labeled probe JVO-0750 and MMO-1056 at 42°C overnight to detect GcvB and 5S rRNA, respectively. The membrane was washed in 5 × SSC/0.1% SDS at 42°C for 15 min, 1 × SSC/0.1% SDS at 42°C for 15 min, and 0.5 × SSC/0.1% SDS at 42°C for 15 min.
The membranes were exposed to imaging plates (Fujifilm), and the resulting signals were visualized using Typhoon FLA7000 scanner (GE Healthcare) and quantified using Image Quant TL software (GE Healthcare).
Growth assay
The E. coli BW25113 ΔgcvBΔsroC strains harboring pBAD-gcvB or its vector control (pKP8-35) were grown overnight in LB medium. The cells were harvested by centrifugation (5,000×g, 20℃, 5 min), washed twice with M9 minimal medium, and diluted to 1:100 in M9 minimal medium supplemented with 0.2% glycerol and 0.2% l-arabinose in the presence or absence of Phe, Tyr, or Trp (final concentration: 1 mM). A 100 µL aliquot of the cell suspension was transferred into 96-well flat-bottom clear microtiter plates (Iwaki). The plates were incubated at 37℃ with rotary shaking at 180 rpm with a 3 mm amplitude for 48 h in Humidity Cassette using Spark plate reader (Tecan). Growth was continuously monitored by measuring OD_600_ every 30 min. Lag time was calculated using microbial lag phase duration calculator (66).
Determination of MIC of azaserine
Biological triplicates of the E. coli BW25113 ΔgcvBΔsroC strains harboring pP_L_-gcvB or its vector control (pTP11) were grown overnight in LB medium. The cells were harvested by centrifugation (5,000×g, 20°C, 5 min), washed twice with M9 minimal medium, and diluted 1:100 in M9 minimal medium supplemented with 0.4% glucose in the presence or absence of azaserine (final concentration: 20–100 μM). A 100 µL aliquot of the cell suspension was transferred into 96-well flat-bottom clear microtiter plates (Iwaki). The plates were incubated at 37°C with rotary shaking at 180 rpm with a 3 mm amplitude for 72 h in Humidity Cassette using Spark plate reader (Tecan). Growth was continuously monitored by measuring OD_600_ every 1 h.
Statistical analysis
The significance of the results was calculated using the two-tailed Student’s t-test or one-way ANOVA with the Bonferroni post hoc test (*P < 0.05, **P < 0.01, ***P < 0.001).
Supplementary Material
Reviewer comments
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ikeda M. 2006. Towards bacterial strains overproducing l-tryptophan and other aromatics by metabolic engineering. Appl Microbiol Biotechnol 69:615–626. doi:10.1007/s 00253-005-0252-y 16374633 · doi ↗ · pubmed ↗
- 2Liu S, Xu J-Z, Zhang W-G. 2022. Advances and prospects in metabolic engineering of Escherichia coli for l-tryptophan production. World J Microbiol Biotechnol 38:22. doi:10.1007/s 11274-021-03212-134989926 · doi ↗ · pubmed ↗
- 3Effendi SSW, Ng I-S. 2023. Challenges and opportunities for engineered Escherichia coli as a pivotal chassis toward versatile tyrosine-derived chemicals production. Biotechnol Adv 69:108270. doi:10.1016/j.biotechadv.2023.10827037852421 · doi ↗ · pubmed ↗
- 4Sprenger GA. 2007. From scratch to value: engineering Escherichia coli wild type cells to the production of l-phenylalanine and other fine chemicals derived from chorismate. Appl Microbiol Biotechnol 75:739–749. doi:10.1007/s 00253-007-0931-y 17435995 · doi ↗ · pubmed ↗
- 5Pittard J, Yang J. 2008. Biosynthesis of the aromatic amino acids. Eco Sal Plus 3:10. doi:10.1128/ecosalplus.3.6.1.826443741 · doi ↗ · pubmed ↗
- 6Akashi H, Gojobori T. 2002. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc Natl Acad Sci U S A 99:3695–3700. doi:10.1073/pnas.06252699911904428 PMC 122586 · doi ↗ · pubmed ↗
- 7Pittard AJ, Davidson BE. 1991. Tyr R protein of Escherichia coli and its role as repressor and activator. Mol Microbiol 5:1585–1592. doi:10.1111/j.1365-2958.1991.tb 01904.x 1943694 · doi ↗ · pubmed ↗
- 8Vitreschak AG, Lyubetskaya EV, Shirshin MA, Gelfand MS, Lyubetsky VA. 2004. Attenuation regulation of amino acid biosynthetic operons in proteobacteria: comparative genomics analysis. FEMS Microbiol Lett 234:357–370. doi:10.1016/j.femsle.2004.04.00515135544 · doi ↗ · pubmed ↗