Diverse Library of 5a-Substituted Carba-Glucosamines
Bjarne Silkenath, Dennis Kläge, Philip Eppelin, Jörg S. Hartig, Valentin Wittmann

TL;DR
Scientists created a diverse set of carba-glucosamine compounds that can act as antibiotics by targeting bacterial cell wall synthesis.
Contribution
A new library of 20 carba-glucosamine derivatives with varied substituents was synthesized and tested for antibacterial activity.
Findings
The compounds were efficiently synthesized from late-stage intermediates.
Some carba-glucosamine derivatives showed promising antibacterial activity against Bacillus subtilis in a filter disk assay.
Abstract
Carba-sugars—carbohydrate mimics in which the ring oxygen is replaced by a methylene group—are carbohydrate analogues of natural or synthetic origin that can have important biological functions. Especially, carba-aminosugars and glycosides containing carba-aminosugars are potent antibiotics. Furthermore, they have been shown to induce the self-cleavage reaction of the glmS riboswitch and thereby inhibit the ability of bacteria to synthesize glucosamine-6-phosphate, which is required to build up the bacterial cell wall. We report the synthesis of a library of 20 carba-glucosamine derivatives with various substituents at the carba-position including amines, alkyl, alkoxy, and aryloxy derivatives, fluorine derivatives, glycosylated derivatives, and a cyclopropane derivative. The compounds were obtained in an efficient way starting from late-stage synthetic intermediates of an…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
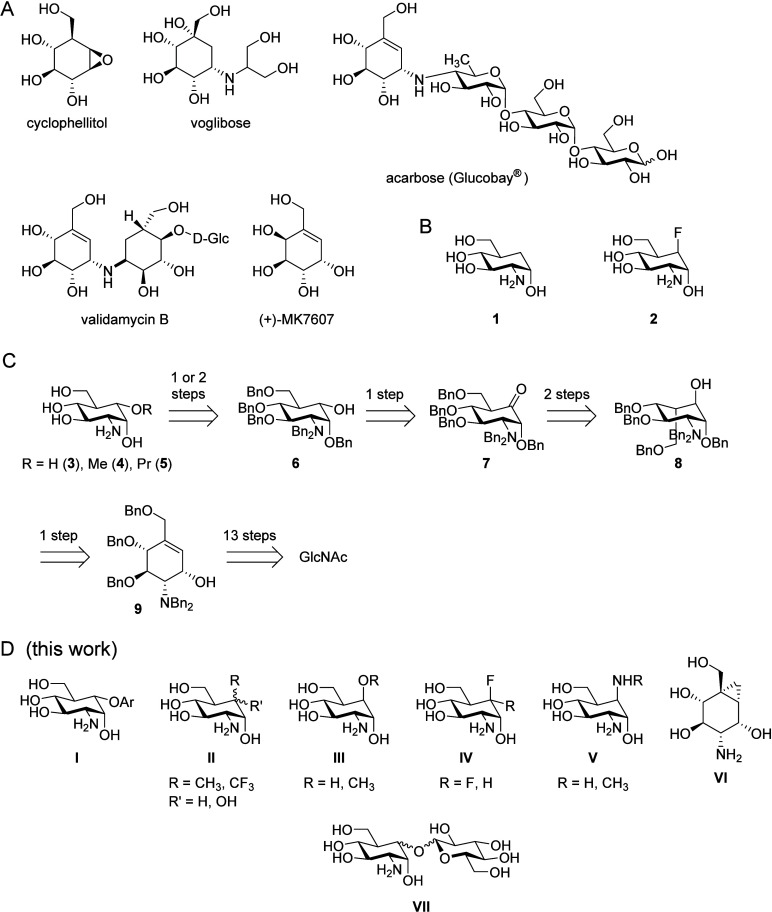 Figure 1
Figure 1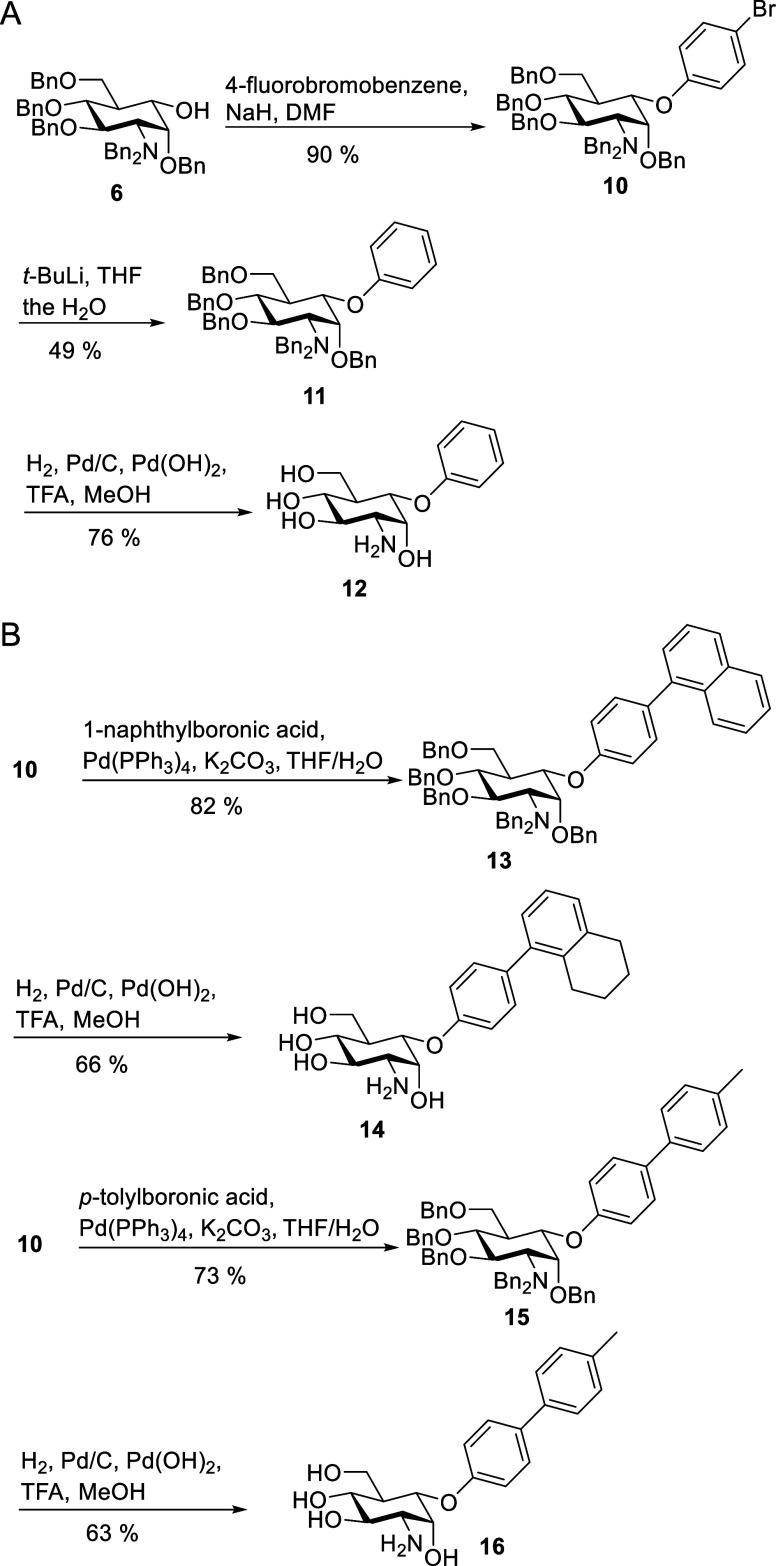 Scheme 1
Scheme 1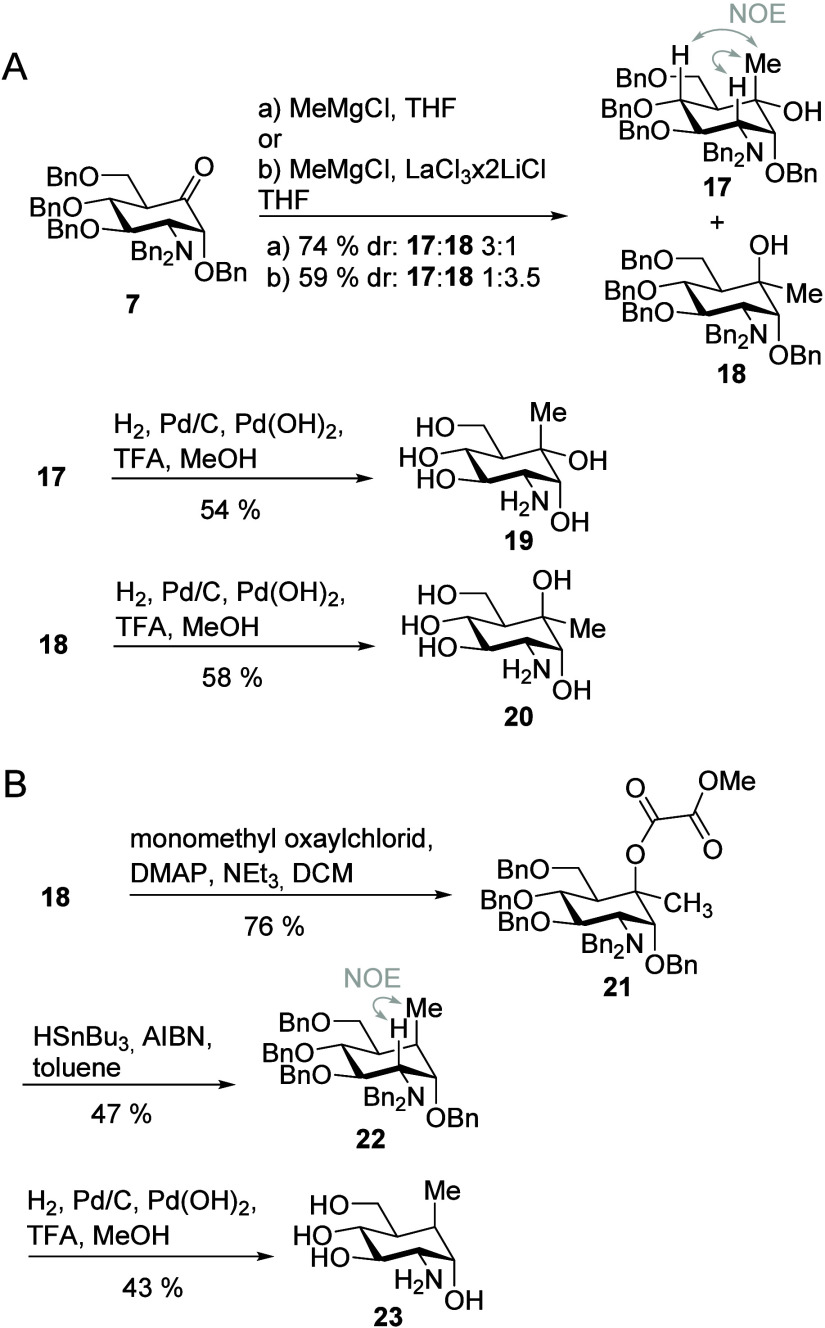 Scheme 2
Scheme 2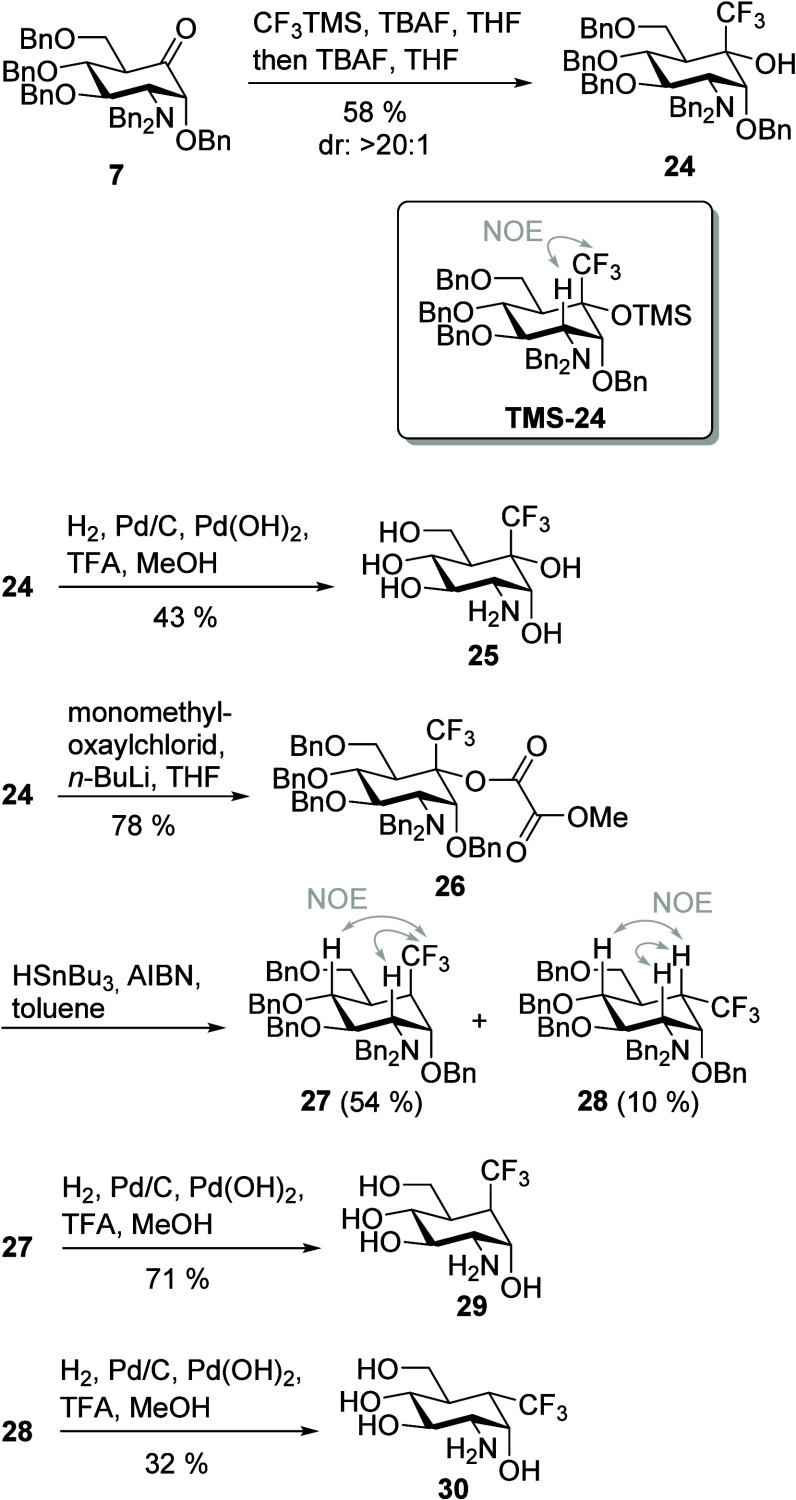 Scheme 3
Scheme 3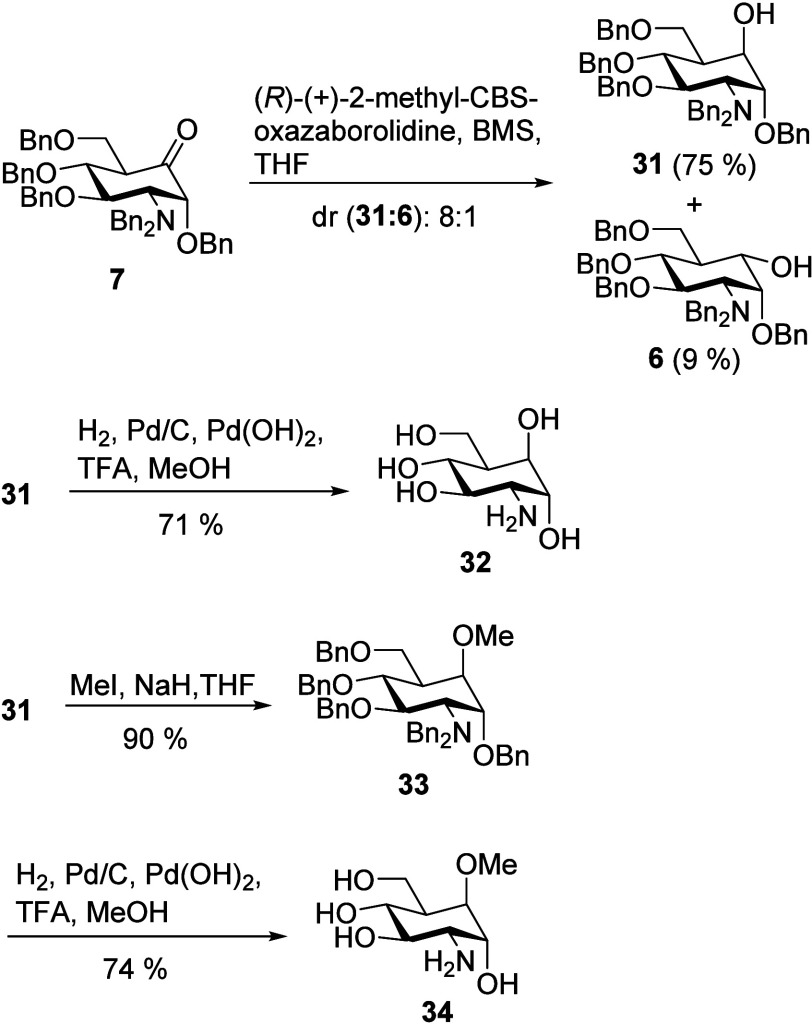 Scheme 4
Scheme 4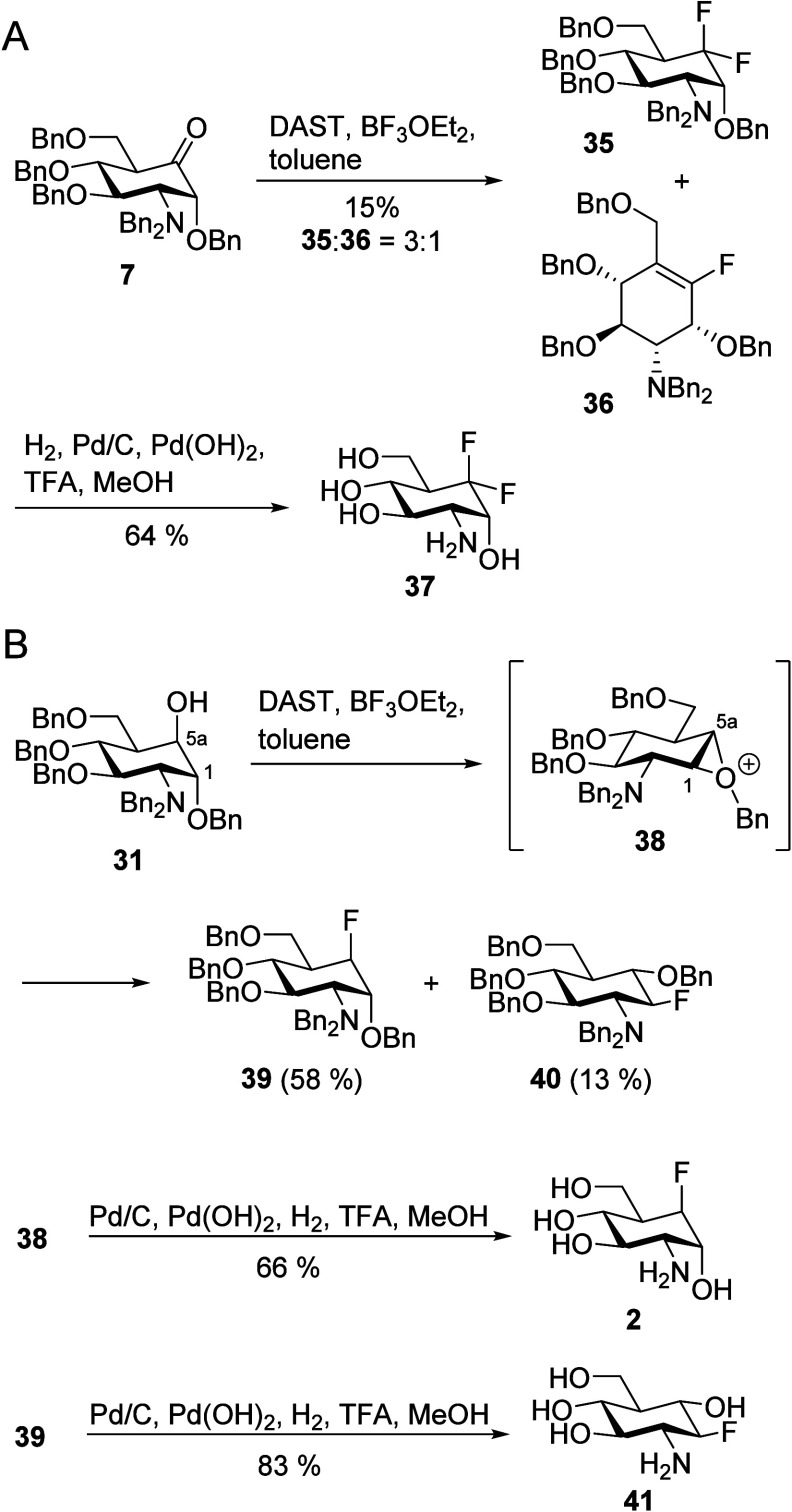 Scheme 5
Scheme 5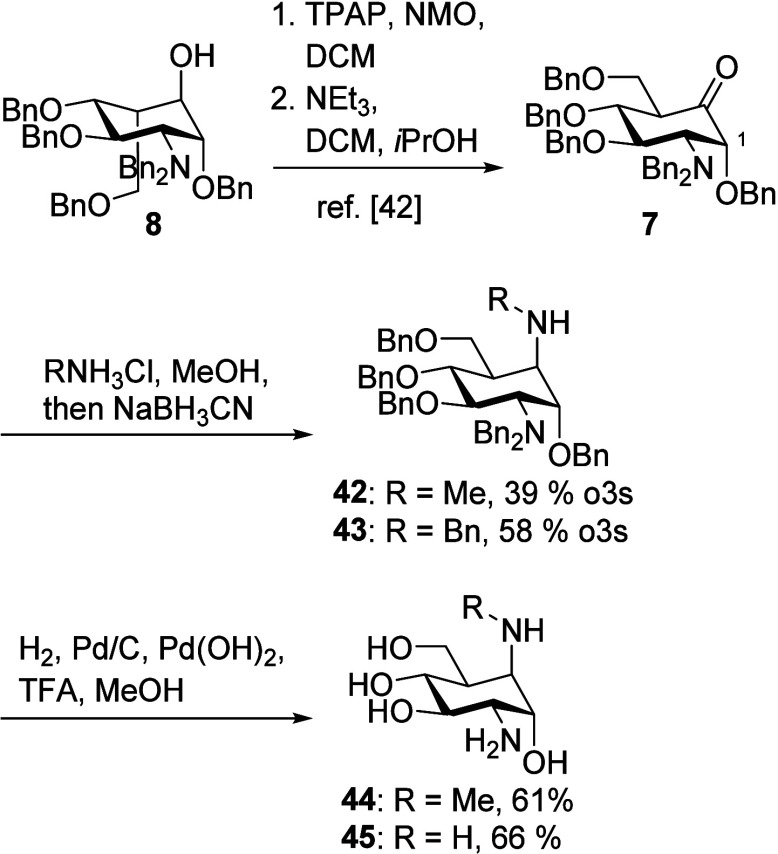 Scheme 6
Scheme 6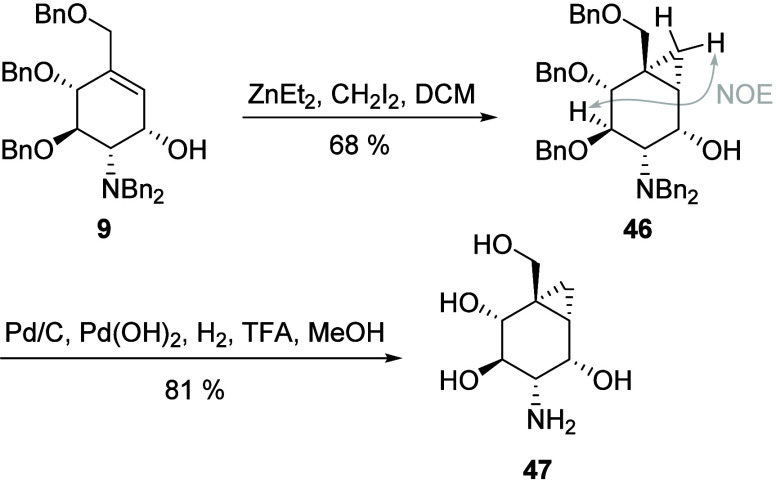 Scheme 7
Scheme 7 Scheme 8
Scheme 8 Figure 2
Figure 2- —Deutsche Forschungsgemeinschaft10.13039/501100001659
- —Konstanz Research School Chemical BiologyNA
- —Universität Konstanz10.13039/501100010583
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCarbohydrate Chemistry and Synthesis · Chemical Synthesis and Analysis · Glycosylation and Glycoproteins Research
Introduction
Carba-sugars are carbohydrate mimics in which the ring oxygen, which is part of the hemiacetal function, is replaced by a methylene group.^1^ This position is termed the 5a position. A multitude of these compounds have been either extracted from nature or synthesized.^2−4^ Many carba-sugars possess interesting biological activities ranging from the inhibition of glycosidases used in part to treat diabetes (cyclophellitol, voglibose, and acarbose)^5−13^ to antimicrobial activity (validamycins and MK7607)^14−17^ (Figure 1A). Especially, carba-aminosugars and glycosides containing carba-aminosugars are potent antibiotics.^3^ Previously, we have shown that the carba-analogue 1 of glucosamine and fluorine derivative 2 possess antibiotic properties (Figure 1B).^18,19^ This was explained by their ability to catalyze the self-cleavage reaction of the glmS riboswitch and thereby inhibit the ability of bacteria to synthesize glucosamine-6-phosphate, which is required to build up the bacterial cell wall.^19,20^ The antibiotic properties of carba-aminosugars 1 and 2 sparked our interest in the synthesis of modified carba-glucosamines to fully assess the scope of these antimicrobial compounds. Especially, the modification of the 5a (carba) position was of interest to us as it offers room for derivatization not available in the parent carbohydrates. Previous synthetic strategies for carba-sugars provided one target structure at the end of the synthesis. These strategies used the Ferrier rearrangement,^21−24^ Diels–Alder chemistry,^25−31^ various aldol reactions,^32−34^ ring-closing metathesis,^35−39^ as well as microbial oxidations of benzene derivatives.^40,41^ To the best of our knowledge, none of these synthetic strategies allowed for selective derivatization of the 5a position unique to carba-sugars.^3,4^ In our previous research, we established for the first time a synthetic route toward carba-analogues 3–5 of glucosamine bearing equatorially oriented alkoxy substituents in the 5a position (Figure 1C).^42^ In light of the multitude of potential biological applications of these compounds, we envisioned the synthesis of a large library of 5a-substituted carba-aminosugars utilizing late-stage intermediates of our established synthesis. Such a library would allow a better understanding of the antibiotic potential of this compound class. The required flexible synthetic strategy would also be of high interest to other fields of research, such as the development of new antidiabetic compounds or glycosidase inhibitors.
(A) Biologically active carba-sugars: glycosidase inhibitors cyclophellitol, voglibose, and acarbose and antimicrobial compounds validamycin B and (+)-MK7607. (B) Carba-glucosamine (carba-GlcN) analogues 1 and 2 with antibiotic activity that have been shown to activate the glmS riboswitch. (C) Retrosynthesis of published 5a-hydroxy- and 5a-alkoxy-modified carba-glucosamine mimics 3–542 with late-stage intermediates that served as starting materials for the carba-glucosamine mimics reported here. (D) Library of new carba-glucosamine mimics reported in this work.
Here, we report the synthesis of a diverse library of 20 carba-glucosamine mimics with various substituents at the 5a position (Figure 1D) making use of late-stage synthetic intermediates of our previously developed synthesis of 5a-substituted carba-glucosamines. Starting from 5a-hydroxy (6, 8), 5a-oxo (7), or cyclohexene (9) derivatives, we employed different reaction types to achieve a variety of compounds with new substitution patterns at C-5a that were previously not accessible. We tested all carba-glucosamine mimics for their antibacterial properties against Bacillus subtilis and discovered promising activities for some compounds.
Results and Discussion
Figure 1C depicts the retrosynthesis of the 5a-hydroxy and -alkoxy derivatives 3–5 that we published earlier.^42^ The synthesis starts with N-acetylglucosamine (GlcNAc) and delivers the target structures in 18–19 steps. Intermediates 6–9 occur at a late stage of the synthesis and therefore serve as ideal starting points for the diversification presented in the current study. We started the investigation of new 5a substituents with the introduction of aryloxy substituents (substitution type I). Reactions for the installation of phenoxy groups starting from an alcohol are rare.^43,44^ We opted for the generation of a bromophenoxy substituent via an S_N_Ar reaction, offering the possibility to remove the bromine to give a phenoxy substituent as well as to access more complex aryl modifications by subsequent cross-coupling reactions. Addition of C-nucleophiles to ketone 7 provided access to carba-glucosamine mimics of type II with 5a mono- and disubstitution. Reduction of 7 with CBS reagents provided carba-GlcN derivatives with substitution type III bearing axial alkoxy or hydroxy substituents. These compounds are stereoisomers of the equatorially substituted compounds 3–5 depicted in Figure 1C. It was further possible to introduce fluorine by fluoride addition to ketone 7 and subsequent optional deoxygenation resulting in substitution type IV. Reductive amination of the carbonyl of 7 yielded substitution type V, a carba-GlcN with an additional amine in the 5a position. A bicyclic carba-GlcN (substitution type VI) was prepared by Simmons–Smith cyclopropanation of 9. It was also possible to glycosylate the hydroxy group in the 5a position of 6 and its epimer III (R = H) to generate a new type of carba-disaccharides (substitution type VII).
5a-Aryloxy-Substituted Carba-Glucosamine Mimics (Substitution
Type I)
Carba-sugars bearing a phenoxy substituent in position 5a are promising antibacterial compounds for several reasons. The aryl moiety might allow them to intercalate in RNA structures, such as the glmS riboswitch, for which glucosamine mimics have been shown to be potent activators.^18−20^ Furthermore, the aryl moiety decreases the polarity of the glucosamine mimic to a point where passive diffusion into the bacterium can be considered reasonable. Ikegai reported the use of tetraphenyl bismuth fluoride for O-phenylation of cyclohexanol under copper catalysis.^43^ However, application of the reported conditions to alcohol 6 gave only trace amounts of the desired phenoxy carba-sugar 11 (Scheme 1A). In an alternative route, alcohol 6 was reacted in an S_N_Ar reaction with 4-fluorobromobenzene^45^ to give compound 10 in a yield of 90%. Compound 10 was successfully dehalogenated by treatment with t-BuLi and aqueous workup to give phenoxy-modified glucosamine mimic 11 in a yield of 49%. This compound was deprotected by hydrogenation using a mixture of Pd(0) on carbon and Pd(OH)2 on carbon as catalysts^46^ in the presence of trifluoroacetic acid to give compound 12 in a yield of 76%. This was achieved without affecting the aryl moiety at position 5a.
(A) Synthesis of Phenyloxy Derivative 12; (B) Synthesis of Aryloxy Derivatives 14 and 16 Obtained by Suzuki–Miyaura Cross-Couplings
The intermediate aryl bromide 10 offered room for further modification. We decided to introduce larger aromatic residues to further decrease the polarity of the target compound and potentially facilitate intercalation into RNA. To achieve this, Suzuki–Miyaura cross-coupling^47^ was performed (Scheme 1B). With 1-naphthylboronic acid, the naphthyl-modified glucosamine 13 was obtained in a yield of 82%. When p-tolylboronic acid was used as the boronic acid, biphenyl compound 15 was isolated in a yield of 73%. When these compounds were subjected to hydrogenation, naphthyl derivative 13 reacted to tetrahydronaphthyl compound 14 in a yield of 66%. Compound 15 was deprotected to obtain 16 in a yield of 63%.
5a-Methyl-Substituted Carba-Glucosamine Mimics (Substitution
Type II)
Carba-glucosamine mimics bearing an additional carbon–carbon bond in the 5a position have not yet been reported. To access this compound class, methyl magnesium chloride was reacted with ketone 7 resulting in a separable 3:1 mixture of compounds 17 and 18 in a yield of 74% (Scheme 2A). The assignment of the configuration at the newly formed stereocenter (position 5a) was possible by NMR spectroscopy. In isomer 17, NOEs between the methyl group and H-2 as well as H-4 could be observed, which were missing in the NOESY spectrum of 18 (Scheme 2A and Supporting Information). The observed stereoselectivity of the attack of the Grignard reagent to this cyclohexanone derivative leading to the preferred formation of an axial methyl group is in accordance with previously published reactions of cyclohexanones.^48^ When adding LaCl_3_x2LiCl^49^ to compound 7 before the addition of the Grignard reagent, an inversion of the stereochemical outcome was observed, and a 1:3.5 mixture of 17 and 18 was obtained in a yield of 59%. A plausible explanation for the inverted stereoselectivity is the complexation of the lanthanide(III) salt to the carbonyl group, blocking the attack from the axial direction. Equatorial attack of organometallic reagents to cyclohexanones is also observed in the presence of CeCl_3_.^50^ The unprotected disubstituted carba-sugars 19 and 20 were obtained in a yield of 54 and 58%, respectively, upon hydrogenation.
(A) Synthesis of 5a-Methyl-5a-hydroxy-Disubstituted Carba-Sugars 19 and 20; (B) Deoxygenation of 18 and Deprotection to Give Carba-Glucosamine 23
Compounds 17 and 18 were expected to be promising starting materials for deoxygenation, allowing access to solely 5a-methyl-modified carba-glucosamine mimics. This approach, however, proved to be challenging, and when xanthates were employed for Barton–McCombie deoxygenation,^51^ only trace amounts of the desired products were observed. Alternative deoxygenation methods reported by Jang et al.^52^ and Yasuda et al.^53^ did not provide access to the methyl carba-glucosamines either. Conversion of tertiary alcohol 18 to methyl oxalic acid ester 21 according to Dolan and MacMillan,^54^ however, succeeded in a yield of 76%. Subsequent radical deoxygenation employing HSnBu_3_ and AIBN allowed for the synthesis of carba-glucosamine 22 in a yield of 47%. Hydrogen transfer from HSnBu_3_ to the intermediate C-5a radical exclusively led to the formation of an axial 5a-methyl group, as proven by a NOESY NMR spectrum. Interestingly, the same sequence starting from 17 proceeded with lower yields and stereoselectivity. Compound 22 was deprotected by hydrogenation to give methyl-modified carba-glucosamine 23 in a yield of 43%.
We next aimed for the introduction of an electron-withdrawing trifluoromethyl group in the 5a-position, which was achieved by treatment of ketone 7 with CF_3_TMS (Ruppert–Prakash reagent)^55^ and catalytic amounts of TBAF. Subsequent addition of a stoichiometric amount of TBAF yielded alcohol 24 in 58% with excellent diastereoselectivity. Assignment of the configuration at the carbinol center (C-5a) was carried out by HOESY NMR analysis of intermediate TMS ether TMS-24, which was isolated in small amounts (see the Supporting Information). Hydrogenation of the benzyl-protecting groups gave compound 25 in a yield of 43% (Scheme 3). Deoxygenation of compound 24 was performed as described for compound 18. Formation of the corresponding methyl oxalic acid ester 26 succeeded in a yield of 78%. Subsequent treatment with HSnBu_3_ and AIBN proceeded with incomplete stereoselectivity to give carba-glucosamine 27 with an axial and 28 with an equatorial CF_3_ group in yields of 54% and 10%, respectively. The stereochemistry at position 5a of 27 and 28 was confirmed by NOESY and HOESY NMR spectra (Scheme 3 and the Supporting Information).
Trifluoromethylation of Ketone 6 Provided Access to 5a-CF3-Substituted Carba-Glucosamines 25, 29, and 30
Axial 5a-Hydroxy and -Alkoxy Substituents (Substitution Type
III)
We previously reported the synthesis of carba-glucosamines with equatorial hydroxy (3) and alkoxy substituents (4, 5).^42^ Compound 3 was obtained from ketone 7 by treatment with (S)-(−)-2-methyl-CBS oxazaborolidine as a catalyst and borane dimethyl sulfide as a reducing agent followed by hydrogenation.^42^ Keeping in mind the huge influence of the stereochemistry of a carbohydrate derivative on its biological function, we wanted to provide access to 5a-modified carba-glucosamines with an axial hydroxy group. To access this diastereomer of 6, we reduced ketone 7 by applying (R)-(+)-2-methyl-CBS oxazaborolidine as a catalyst providing compound 31 in a yield of 75% beside small amounts of the diastereoisomer 6 with an equatorial hydroxy group (Scheme 4). Deprotection of 31 gave 5a-axially modified carba-glucosamine mimic 32. To obtain methoxy derivative 33, 31 was methylated with methyl iodide in a yield of 90%. Debenzylation of methyl ether 33 finally provided carba-glucosamine mimic 34 with an axial methoxy group.
Preparation of Axial Alcohol 31 by CBS Reduction of Ketone 7 and Further Reaction to Carba-Sugars 33 and 34
5a-Fluoro-Substituted Carba-Glucosamine Mimics (Substitution
Type IV)
Fluoro-modified carba-glucosamine 2 has been shown to have promising antimicrobial activity.^18^ Furthermore, Deleuze et al. reported the synthesis of gem-difluoro-carba-glucose via a triisobutylaluminum-promoted rearrangement^56^ and found that the difluoro substitution restores the anomeric effect.^57^ This implies that gem-difluoro-carba-glucosamine 37 might be a very promising mimic of glucosamine, and therefore, we attempted to extend the sugar mimic library toward this compound. Geminal difluorides can be generated from ketones by reaction with DAST.^58,59^ For ketones with a tertiary α position, however, this kind of conversion has been reported to be difficult, suffering from rearrangement and elimination side reactions.^59,60^ Indeed, when we treated ketone 7 with DAST, we first observed only traces of difluoride 35 by LC-MS and ^19^F NMR in a very complex product mixture. After extensive optimization of the conditions, 10 equiv of DAST and 1 equiv of boron trifluoride diethyl etherate in toluene in a PTFE reaction vessel gave 15% of an inseparable 3:1 mixture of difluoride 35 and the elimination product 36 (Scheme 5A). After hydrogenation, deprotected difluoride 37 was isolated in a pure form by HILIC-HPLC.
(A) Difluorination of Ketone 7 with DAST Resulted in a Mixture of 35 and 36; (B) Deoxyfluorination of Axial Alcohol 31 Afforded a Mixture of 39 (Major Product) and Rearrangement Product 40The mixture was hydrogenated upon which pure 37 could be separated.Hydrogenation of the separated compounds gave 2 and 41, respectively.
Previous approaches to the monofluorinated carba-glucosamine analogous to compound 2 but with the fluoride in equatorial position were not successful.^18^ Carba-sugar 2 was synthesized by a completely different approach relying on a Ferrier rearrangement and introduction of the fluoride via electrophilic fluorination in the α position to the resulting ketone.^18^ To provide access to the equatorial fluoride, we aimed for the conversion of the axial alcohol 31 to the fluoride in an S_N_2 fashion by deoxyfluorination. Surprisingly, this reaction yielded two compounds (Scheme 5B). The major product was identified as the axial fluoride 39, which was obtained in a yield of 58% beside rearrangement product 40 (13%). The formation of these two compounds hints at the formation of oxonium ion intermediate 38, which is formed by a neighboring group effect. Opening of 38 by fluoride results either in 39 under retention of configuration at the 5a position (^3^JH-5,F = 37 Hz) via double inversion (major product according to the Fürst–Plattner rules) or in 1-fluoro compound 40 (^1^H NMR signal of H-1: 4.75 ppm, ddd, ^2^JH,F = 51.4 Hz, ^3^JH,H = 10.4, 8.7 Hz) under migration of the benzyl ether to the 5a-position. Both compounds were isolated and deprotected by hydrogenation to yield compounds 2 and 41, respectively. Even though the synthesis of fluoro carba-sugar 2 starting from glucosamine hydrochloride has been published,^18^ the synthetic approach presented here starting from GlcNAc is much more efficient with an overall 25-fold improvement of the total yield.
5a-Amine-Substituted Carba-Glucosamine Mimics (Substitution
Type V)
The carba-glucosamine derivatives 1 and 2 have been shown to activate the self-cleavage reaction of the glmS riboswitch. The first step of this process is binding of the carba-glucosamine derivative to the riboswitch. Since RNA is a polyanionic molecule, multiple amino groups can be beneficial for binding to RNA. Indeed, prime examples of naturally occurring RNA ligands are aminoglycosides, which are carbohydrate-like structures with several amino groups.^61^ To potentially increase the RNA affinity of carba-glucosamine derivatives, we aimed at the introduction of an additional amino group in the 5a position. Ley–Griffith oxidation of 8 and subsequent isomerization following our previously published procedure^42^ gave ketone 7, which was reacted as the crude product in a reductive amination using methyl or benzyl ammonium chloride and sodium cyanoborohydride as the reducing agent yielding diamines 42 and 43, respectively (Scheme 6). NMR analysis revealed that the introduced amine was exclusively in the axial position, which is in line with our hypothesized coordination of cyanoborohydride to the benzyl ether in the 1-position. This precomplexation was expected to result in the introduction of the hydride from the equatorial position. The configurational assignment was based on the observed multiplicity of H-5a (pseudo-triplet with J = 3.2 Hz), indicating the absence of an axial–axial coupling and, therefore, its equatorial orientation. Compounds 42 and 43 were deprotected by catalytic hydrogenation with a mixture of Pd(0) on carbon and Pd(OH)2 on carbon, resulting in the amino-modified carba-glucosamine analogues 44 and 45 in yields of 61 and 66%, respectively.
Synthesis of the 5a-Amino-Substituted Carba-Glucosamines 44 and 45
Bicyclic Carba-Glucosamine Mimics (Substitution Type VI)
Apart from the above-mentioned intermediates of our previously developed carba-sugar synthesis, allyl alcohol 9 provided another point of modification. Allylic alcohol 9, which was the product of a ring-closing metathesis reaction, proved to be ideal for a diastereoselective Simmons–Smith cyclopropanation^62^ that delivered bicyclic carba-glucosamine mimic 46 in a yield of 68% (Scheme 6). A cross peak between H-3 and one of the diastereotopic cyclopropyl protons in a NOESY NMR spectrum confirmed the stereochemistry depicted in Scheme 7. The cyclopropane ring was stable under the hydrogenation conditions needed for benzyl deprotection, and compound 47 was isolated in a yield of 81%.
Synthesis of Bicyclic Carba-Glucosamine 47
5a-Carba-Disaccarides (Substitution Type VII)
To fully show the scope of possible 5a-modifications, glycosylation of the 5a-hydroxy carba-glucosamine derivatives 6 and 31 to form pseudodisaccharides was attempted (Scheme 8). The obtained 1,5a-linked compounds represent interesting new structural motifs, not previously described in the literature. Glycosylation of the equatorial hydroxy group of 6 turned out to be very challenging. A series of glycosyl donors with varying anomeric leaving groups (acetate, activated by BF_3_ etherate or TMSOTf; bromide, activated by AgNO_3_, AgOTf, or Ag_2_CO_3_; phenyl thiolate, activated by N-iodosuccinimide; and trichloroacetimidate, activated by BF_3_ etherate) gave only trace amounts of product. However, reaction of alcohol 6 with 2-O-acetyl-protected glucosyl fluoride 48(63) and BF_3_ etherate as an activator gave β-1,5a-pseudodisaccharide 49 in a yield of 46%. The neighboring group effect of the acetyl-protecting group ensured a high β-selectivity of α/β 1:10 of the glycosylation reaction. The configuration at the anomeric center of the glucose moiety was confirmed by the H1′–H2′ coupling constant (^3^J = 7.9 Hz). Deacetylation with potassium carbonate in MeOH gave compound 50 in a quantitative yield. Subsequent hydrogenation removed the benzyl-protecting groups to obtain unprotected β-1,5a-pseudodisaccharide 51 in a yield of 75%. When the equatorial 5a alcohol 6 was reacted with perbenzylated glycosyl fluoride 52,^64^ α-product 53 (^3^JH1′,H2_′_ = 3.5 Hz) was obtained with high selectivity (α/β 15:1) in a yield of 41%. Compound 53 was debenzylated to give α-1,5a-pseudodisaccharide 54. Reaction of the axial 5a alcohol 31 with fluoride 52(64) gave α-product 55 (^3^JH1′,H2_′_ = 3.6 Hz) in a yield of 44%. 55 could be successfully deprotected by hydrogenation to provide α-1,5a-pseudodisaccharide 56. These experiments demonstrate the high degree of stereochemical control that can be achieved at the obtained 1,5a-glycosidic bond.
Preparation of Pseudo-Disaccharides 51, 54, and 56
Antibacterial Assays
All synthesized carba-glucosamine mimics were tested for their antimicrobial properties in filter disk assays. Glucosamine (GlcN) was used as the negative control. This carbohydrate is a substrate of phosphotransferase transporter systems, which couple active transport over the bacterial membrane with phosphorylation of the 6-hydroxy group, thereby producing natural, nontoxic GlcN6P. The known antibiotic chloramphenicol (Cm) served as a positive control. For the monofluoro-substituted carba-glucosamine 2, we observed growth inhibition of B. subtilis, which is in line with the previously published results. In addition, we observed clear inhibition zones for the 5a-aryloxy-substituted carba-glucosamines 14 and 16, whereas phenyloxy-substituted carba-glucosamine 12 did not show any growth inhibition (Figure 2). All other compounds did not show an inhibitory effect, as well.
Filter disk assay on Luria–Bertani (LB) agar plates. B. subtilis wt 168 was plated out and tested with 10 μL of a 100 mM solution of the respective compound on a filter disk. Cm: chloramphenicol at a concentration of 9.3 mM.
Conclusions
In this publication, we report the synthesis of a diverse library of new unnatural carba-glucosamine mimics with various substituents in the 5a-position. Starting from late-stage intermediates of a previously published synthesis of carba-glucosamines, it was possible to access aryloxy-, alkyloxy-, alkyl-, amine-, and fluoro-substituted as well as disubstituted carba-sugars, pseudodisaccharides, and a bicyclic derivative. The reported synthesis is flexible, giving access to a large variety of 5a-modified carba-α-d-glucosamines in reasonable yields and with a controllable stereochemical outcome. Given the fact that many naturally occurring carba-sugars have interesting biological activities, we tested these new members of this still “juvenile” compound class of substituted carba-sugars for their antimicrobial potential. The aryl-modified compounds 14 and 16 showed growth inhibition of B. subtilis and, therefore, can be considered as a starting point for the development of new antibiotics. We are convinced that the synthesis of this library is not only of interest for antimicrobial research but also will spark the attention of colleagues working on other applications of carbohydrate mimics, such as antidiabetic compounds or glycosyltransferase inhibitors.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mc Casland G. E.; Furuta S.; Durham L. J. Alicyclic Carbohydrates. XXIX. The Synthesis of a Pseudo-Hexose (2,3,4,5-Tetrahydroxycyclohexanemethanol). J. Org. Chem. 1966, 31 (5), 1516–1521. 10.1021/jo 01343 a 048. · doi ↗
- 2Suami T.; Ogawa S. Chemistry of Carba-Sugars (Pseudo-Sugars) and their Derivatives. Adv. Carbohydr. Chem. Biochem. 1990, 48, 21–90. 10.1016/S 0065-2318(08)60031-1.2077870 · doi ↗ · pubmed ↗
- 3Arjona O.; Gómez A. M.; López J. C.; Plumet J. Synthesis and Conformational and Biological Aspects of Carbasugars. Chem. Rev. 2007, 107 (5), 1919–2036. 10.1021/cr 0203701.17488060 · doi ↗ · pubmed ↗
- 4Zorin A.; Klenk L.; Mack T.; Deigner H.-P.; Schmidt M. S. Current Synthetic Approaches to the Synthesis of Carbasugars from Non-Carbohydrate Sources. Top. Curr. Chem. 2022, 380 (2), 1210.1007/s 41061-022-00370-0.PMC 882741135138497 · doi ↗ · pubmed ↗
- 5Chen X.; Zheng Y.; Shen Y. Voglibose (Basen, AO-128), one of the most important α-glucosidase inhibitors. Curr. Med. Chem. 2006, 13 (1), 109–116. 10.2174/092986706789803035.16457643 · doi ↗ · pubmed ↗
- 6Chen X.; Lu Y.; Fan Y.; Shen Y., Chapter 5 - Voglibose: An Important Drug for Type 2 Diabetes. In Validamycin and its Derivatives; Chen X.; Lu Y.; Fan Y.; Shen Y., Eds.; Elsevier: 2017; pp 237–278.
- 7Schmidt D. D.; Frommer W.; Junge B.; Müller L.; Wingender W.; Truscheit E.; Schäfer D. alpha-Glucosidase inhibitors. New complex oligosaccharides of microbial origin. Naturwissenschaften 1977, 64 (10), 535–536. 10.1007/BF 00483561.337162 · doi ↗ · pubmed ↗
- 8Coniff R.; Krol A. Acarbose: a review of US clinical experience. Clin. Ther. 1997, 19 (1), 16–26. 10.1016/S 0149-2918(97)80069-0.9083705 · doi ↗ · pubmed ↗