Multiple Desmoplastic Small Round Cell Tumor in the Intestine: A Case Report
Naoto Tsujimura, Mitsuyoshi Tei, Daisuke Umeda, Koki Ishimaru, Shoko Minamiura, Takehiro Yamamoto, Soichiro Mori, Kentaro Nishida, Yukihiro Yoshikawa, Masatoshi Nomura, Koki Tamai, Takuya Hamakawa, Daisuke Takiuchi, Hironao Yasuoka, Masanori Tsujie, Yusuke Akamaru

TL;DR
This case report describes a rare desmoplastic small round cell tumor in the intestine of a 38-year-old male, diagnosed after surgery and confirmed through histopathology and genetic analysis.
Contribution
The paper presents a rare clinical case of DSRCT in the intestine, emphasizing the need for more case reports to improve treatment strategies.
Findings
The patient had multiple tumors in the descending and sigmoid colon with peritoneal dissemination.
Histopathology and genetic analysis confirmed the diagnosis of desmoplastic small round cell tumor.
The patient underwent successful surgery and will continue chemotherapy.
Abstract
Desmoplastic small round cell tumor (DSRCT) is a highly malignant sarcoma and an extremely rare tumor, predominantly found in the abdominal and pelvic regions. Here, we report the case of a patient who underwent surgical treatment for multiple desmoplastic round cell tumor in the intestine. A 38-year-old male patient visited our hospital after a health check revealed positive occult blood in his stool and a colonoscopy revealed tumors in descending colon and sigmoid colon. Biopsy results revealed poorly differentiated adenocarcinoma. Chest and abdominal enhanced computed tomography revealed 3 tumors from descending colon to sigmoid colon and numerous peritoneal disseminations. Based on these findings, we diagnosed multiple colon cancers and performed a laparoscopic left hemicolectomy. Hematoxylin–Eosin (H&E) staining showed that in all tumors, atypical cells with large and small…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
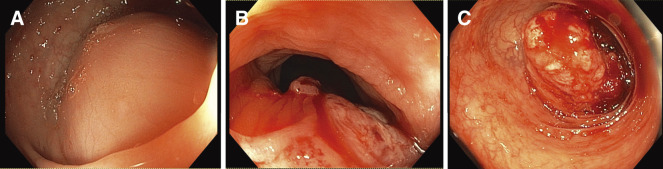 Fig. 1
Fig. 1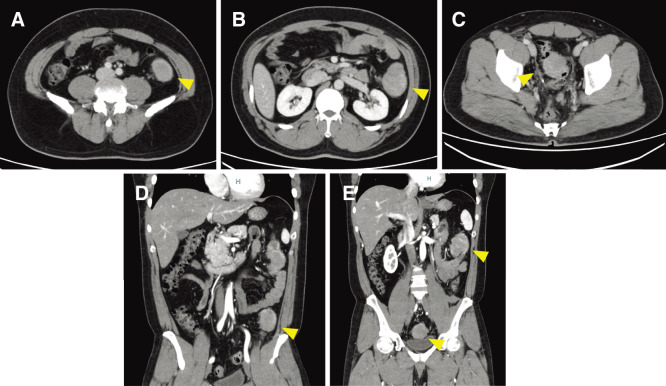 Fig. 2
Fig. 2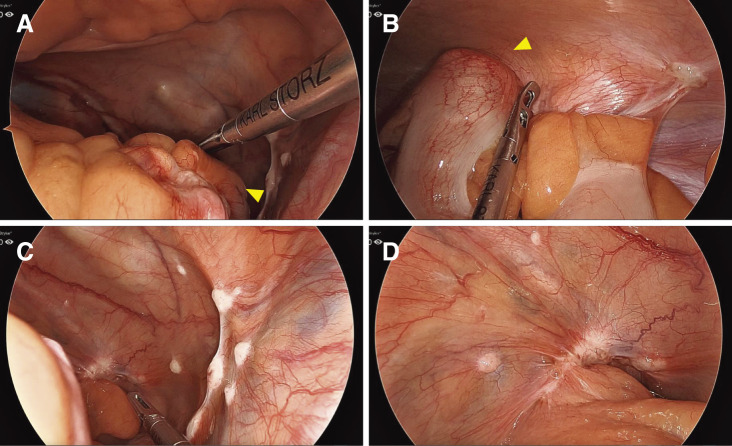 Fig. 3
Fig. 3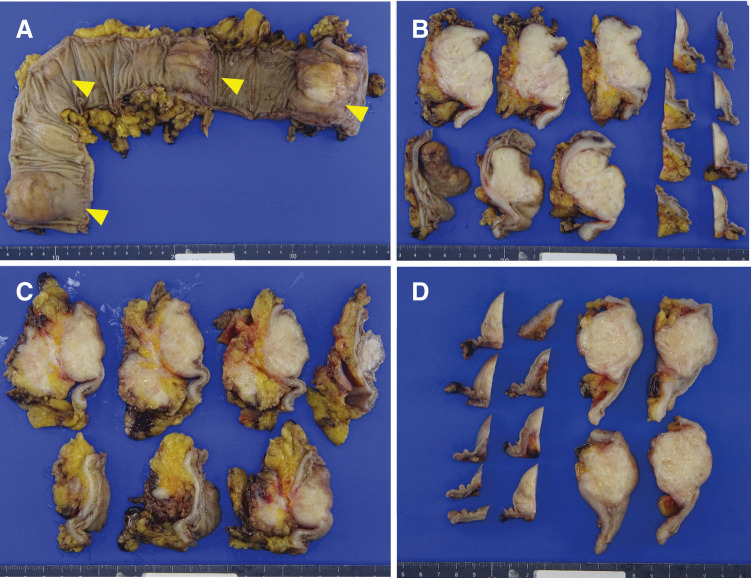 Fig. 4
Fig. 4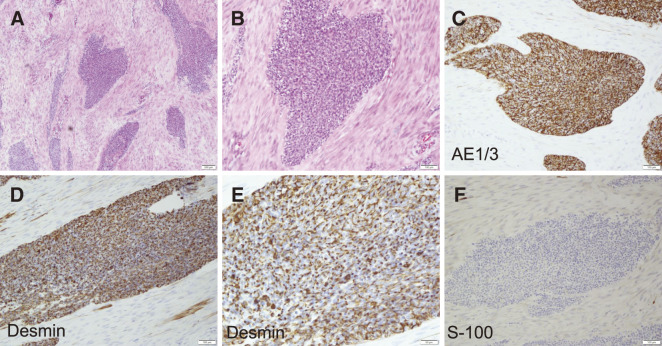 Fig. 5
Fig. 5| Item | Results | Units |
|---|---|---|
| Routine blood test | ||
| WBC | 5.47 | 103/μL |
| NEU | 3.047 | 103/μL |
| NEU-R | 55.7 | % |
| LYM | 1.849 | 103/μL |
| LYM-R | 33.8 | % |
| HGB | 13.8 | g/dL |
| PLT | 22.6 | 104/μL |
| Biochemistry test | ||
| ALT | 54 | U/L |
| AST | 32 | U/L |
| T-BIL | 0.5 | mg/dL |
| LDH | 129 | U/L |
| ALP | 104 | U/L |
| Urea | 10 | mg/dL |
| Crea | 0.8 | mg/dL |
| Serum tumor markers | ||
| CEA | 2.6 | ng/mL |
| CA19-9 | 6 | U/mL |
| Authors | Age/Sex | Surgery | Metastasis before operation | Additional therapy after operation | Recurrence | Recurrence | Time to recurrence | Alive or dead | Time to death |
|---|---|---|---|---|---|---|---|---|---|
| Chen et al. | 18/M | Exploratory laparotomy | Peritoneum, liver, pleura, bone, muscle | ND | Dead | NS | |||
| Laurens et al. | 56/F | En-block excision of tumor | Peritoneum | ND | No | NS | |||
| Pahuja et al. | NS/M | En-block excision of tumor | Peritoneum | P6 regimen and RT | No | Dead | 22 | ||
| Huang et al. | 30/F | Transverse colectomy | No | ND | No | NS | |||
| Liu et al. | 14/F | PD | Lymph nodes | Carboplatin and duoxitasai | Yes | Intraperitoneal | 2 | Dead | 6 |
| Present case | 38/M | Laparoscopic left hemicolectomy | Peritoneum | Alive |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSarcoma Diagnosis and Treatment · Soft tissue tumor case studies · Neurofibromatosis and Schwannoma Cases
Abbreviations
AE1/3 Cytokeratin-multi (AE1/3) ALP alkaline phosphatase ALT alanine aminotransferase AR-ASO androgen receptor-directed antisense oligonucleotides AST aspartate aminotransferase CA125 carbohydrate antigen 125 CEA carcinoembryonic antigen CK Cytokeratin CNA copy number alterations CRP C-reactive protein CRS cytoreductive surgery CT computed tomography c-MYC Myc proto-oncogene protein DDR DNA damage repair DST double stapling technique DSRCT desmoplastic small round cell tumor EGFR epidermal growth factor receptor EMA epithelial membrane antigen EMT epithelial–mesenchymal transition EWS Ewing sarcoma EWSR1 Ewing’s sarcoma breakpoint region1 FGFR fibroblast growth factor receptor H&E hematoxylin-Eosin HGB hemoglobin HIPEC hyperthermic intraperitoneal chemotherapy IGF-1 insulin-like growth factor-1 LDH lactate dehydrogenase LYM lymphocyte LYM-R lymphocyte rate MErT mesenchymal–epithelial reverse transition MRI magnetic resonance imaging NEU neutrophil NEU-R neutrophil rate NSE Neuron-specific enolase PDGF platelet-derived growth factor PET positron emission tomography PLT platelet RTK receptor tyrosine kinase SD Sigmoid-Descending colon SMT submucosal tumor T2WI T2 weighted image T-BIL total bilirubin WBC white blood cell WT1 Wilms tumor
INTRODUCTION
Desmoplastic small round cell tumor (DSRCT) is a highly malignant sarcoma and an extremely rare tumor predominantly found in the abdominal and pelvic regions.^1–4)^ Initially described by Gerald and Rosai in 1989, it was officially named in 1991.^5–7)^ DSRCT accounts for <1% of all soft tissue sarcomas and is more prevalent in men, with a peak incidence in the 20s. DSRCT is characterized by small round cells of various sizes within a densely proliferating fibrous stromal matrix.^8)^ The diagnosis is confirmed by detection of the specific chromosomal translocation t (11;22) (p13;q12). This translocation results in an active fusion protein involving the Ewing sarcoma (EWS) and Wilms tumor (WT1) genes and is pathognomonic for the diagnosis.^9)^ DSRCT can metastasize early and recur rapidly despite treatment. Thus, the prognosis is very poor. Despite multimodal treatment based on chemotherapy, surgery, and radiation therapy, durable remission remains rare. We report a case of DSRCT that is a highly malignant sarcoma and extremely rare.
CASE PRESENTATION
A 38-year-old male patient visited our hospital in June 2024. A health check revealed positive occult blood in his stool, and a colonoscopy revealed tumors in the descending colon and sigmoid colon. Physical examination revealed that the patient was in good general condition, had no abdominal pain, and no palpable masses. Blood test findings revealed a white blood cell count of 5470/μL, hemoglobin of 13.8 g/dL, C-reactive protein (CRP) of 0.2 mg/dL, aspartate aminotransferase (AST) of 32 U/L, and alanine aminotransferase (ALT) of 54 U/L. These findings indicated mild liver dysfunction. Tumor marker assessment revealed carcinoembryonic antigen (CEA) of 2.6 ng/mL and CA19-9 of 6 U/mL, both within the normal range (Table 1).
A colonoscopy revealed tumors in the descending colon and sigmoid colon, preventing the passage of the endoscope (Figs. 1A–1C). Biopsy results revealed poorly differentiated adenocarcinoma. Chest and abdominal enhanced computed tomography (CT) revealed 3 tumors from the descending colon to the sigmoid colon (Figs. 2A–2E). No lymph node metastasis was found, but numerous peritoneal disseminations were found. No lung metastasis was found.
Preoperative colonoscopy. (A) The SMT-like tumor was located in the descending colon. The endoscope could not pass through. (B) The tumor with mucosal erythema was located at the SD Junction. The endoscope was able to pass through. (C) The tumor was located in the sigmoid colon, and the mucosa was red and prone to bleeding, but the endoscope was able to pass through.SD, Sigmoid-Descending colon; SMT, submucosal tumor
An abdominal contrast CT scan. Three tumors are detected in the descending colon and sigmoid colon. (A–C) Axial views. (D, E) coronal views. Arrowhead: a lead point. (A, D) The tumor was located at the SD Junction. (B–D) The tumors were located in the descending colon and sigmoid colon.CT, computed tomography; SD, Sigmoid-Descending colon
Based on these findings, we diagnosed multiple colon cancers (descending and sigmoid colon cancer) and performed a laparoscopic left hemicolectomy to control the bleeding because of hematochezia. The intraoperative finding was that tumors were in splenic flexure, Sigmoid-Descending colon (SD) junction, and rectosigmoid (Figs. 3A and 3B). The tumor in the SD junction invaded the abdominal wall, and peritoneal dissemination was found in the Douglas’ pouch (Figs. 3C and 3D). Peritoneal dissemination was not resected. Ascites and liver metastasis were not observed. Reconstruction was selected for double stapling technique (DST) anastomosis using CDH29 (Ethicon, Tokyo, Japan), and colostomy was not created.
Intraoperative findings. (A) The tumor is exposed to the serosal surface and located in splenic flexure (arrowhead, a lead point). (B) Tumor located in SD junction (arrowhead, a lead point). (C, D) Peritoneal dissemination was found in the Douglas’ pouch.SD, Sigmoid-Descending colon
An excised specimen revealed that 4 tumors were found in the resected intestine. All tumors were submucosal tumor (SMT)-like mucosal protrusions, and the cut surfaces of all tumors revealed white solid masses (Figs. 4A–4D). Hematoxylin**–eosin (H&E) staining showed that in all tumors, atypical cells with large and small swollen nuclei formed irregular solid nests of various sizes against a background of extensive desmoplastic or myxomatous stroma (Figs. 5A and 5B**). No lymph node metastasis was detected.
Resected specimen. (A) Resected specimen revealed 4 SMT-like tumors. Arrowhead: a lead point. (B–D) The cut surfaces of all tumors revealed white solid masses. (B, sigmoid colon; C, D, descending colon.)SMT, submucosal tumor
H&E staining and immunohistochemistry findings of excised specimen. (A) H&E staining (×50), (B) H&E staining (×100), (C) AE1/3 (×100), (D) Desmin (×100), (E) Desmin (×200), and (F) S-100 (×100).AE1/3, Cytokeratin-multi (AE1/3); H&E, Hematoxylin–eosin
Immunohistochemistry showed that tumor cells were CAM5.2 (+), AE1/3 (+), Ber-EP4 (–), EMA (+), CD45 (+), NSE (+), Synaptophysin (–), Chromogranin A (–), CD56 (+), NKX2.2 (–), Vimentin (+), GFAP (–), S-100 (–), Desmin (–), SMA (–), WT1 (–), MyoD1 (–), Myogenin (–) (Figs. 5C–5F). Genetic analysis detected the EWS-WT1 fusion gene at another inspection agency. Based on these pathological results, we diagnosed DSRCT.
The patient was discharged on the 19th postoperative day without postoperative complications. Because the case was unresectable with peritoneal dissemination, we decided it was necessary to undergo multidisciplinary treatment, including chemotherapy, and treatment was continued at another hospital.
DISCUSSION
DSRCT is an aggressive soft-tissue sarcoma as a specific disease first described by Gerard and Rosai in 1989 and officially named in 1991.^5–7)^
Generally, DSRCT occurs in the peritoneal cavity, retroperitoneum, and pelvis, but 6% of DSRCT can occur in an extra-abdominal location and can also be found in the lungs, eyes, and salivary glands.^10–12)^ The origin of DSRCT is unclear. Because most patients develop DSRCT within cavities lined with mesothelial cells and because tumor cells show positive immunohistochemical reactions for epithelial and mesenchymal antigens, such as Desmin and WT1, DSRCT is speculated to originate from mesothelial or submesothelial cells.^13)^ The incidence of DSRCT does not differ significantly between races, but it is predominantly male, with a male-to-female ratio of approximately 3–5:1, and it tends to occur more frequently in younger people.^14)^ DSRCT is characterized by a poor prognosis with 15% overall survival at 5 years.^15)^
DSRCT does not have specific clinical symptoms. Most patients present with early symptoms such as abdominal pain, constipation, ascites, and vomiting.^2–4,14,16)^ Due to the serosal spread of DSRCT, Hayes Jordan, and colleagues at MD Anderson Cancer Center in Houston,^4,14)^ Texas established a new staging system:
Stage 1: Patients with limited disease, localized to 1 or 2 sites in the abdomen or 1 site elsewhere.Stage 2: Patients with any amount of extensive peritoneal disease.Stage 3: Patients with liver metastasis and peritoneal disease.Stage 4: Including peritoneal and liver disease as well as disease outside the abdominal cavity and lymph nodes.
However, this proposed staging system has not yet been validated. DSRCT is very difficult to diagnose preoperatively. Tumor markers such as carbohydrate antigen 125 (CA125) and Neuron-specific enolase (NSE) may be elevated, but there are currently no specific tumor markers.^17)^
Although DSRCT has no special imaging characteristics, CT is the most useful initial imaging test, and magnetic resonance imaging (MRI) is useful in diagnosing pelvic and hepatic lesions. Abdominal CT scans often reveal large, heterogeneous tumors in the abdominal cavity and pelvic peritoneum. Ultrasound often reveals low signal intensity. MRI often shows high signal intensity on T2 weighted image (T2WI) and iso-signal intensity on T1WI^.18,19)^ Also, positron emission tomography (PET)-CT may monitor residual disease and provide early detection of recurrence or tumor progression.^20)^
Histological examination and immunohistochemical staining are essential for differential diagnosis. The differential diagnosis of DSRCT can be with a variety of other round-cell tumors, including Ewing sarcoma, rhabdomyosarcoma, small-cell carcinoma, and mesothelioma.^21,22)^ In most cases of DSRCT, the expression of Desmin, Cytokeratin (CK), Epithelial membrane antigen (EMA), and vimentin is positive. Positivity of both Desmin and CK is considered a specific immunodiagnostic indicator of DSRCT. Vimentin positivity suggests that the tumor is derived from myofibroblasts. Such tumors may express epithelial, mesenchymal, neuroendocrine, and other immunophenotypes and have specific cytogenetic profiles.^23,24)^ DSRCT is distinguished by the t (11;22) (p13;q12) chromosomal translocation involving the fusion of the transcriptional activation domain of Ewing’s sarcoma breakpoint region1 (EWSR1) with the WT1 gene.^2)^ Studies have also suggested that the EWSR1-WT1 fusion protein can induce the expression of platelet-derived growth factor A, leading to fibroblast growth and proliferation and the production of a collagenous stroma, which may explain the characteristic reactive fibrosis of DSRCT.^25)^ The EWSR1-WT1 gene fusion forms a chimeric protein acting as a transcription factor with at least 35 known target genes, including platelet-derived growth factor (PDGF),^25)^ insulin-like growth factor-1 (IGF-1) receptor, epidermal growth factor receptor (EGFR), and others such as Myc proto-oncogene protein (c-MYC) and fibroblast growth factor receptor (FGFR). This translocation and the resulting transcriptional changes are believed to be the major drivers in DSRCT^.26)^ Investigation of somatic mutations, copy number alterations (CNA), and chromosomes in DSRCT samples suggested that deregulation of mesenchymal–epithelial reverse transition (MErT)/epithelial–mesenchymal transition (EMT) and DNA damage repair (DDR) may be important in DSRCT.^27)^
There is no established optimal treatment for DSRCT. Currently, multimodal therapy combining chemotherapy, aggressive cytoreductive surgery, adjuvant radiotherapy, and hyperthermic intraperitoneal chemotherapy (HIPEC) is considered the standard of care for patients without extraperitoneal metastases.^28–30)^ As most cases present as intraperitoneal tumors, the main guidelines recommend initiating treatment with systemic chemotherapy.
Surgery for DSRCT has been reported to be beneficial. Hassan et al. reported that patients who underwent surgical resection had a median survival of 34 months compared with 14 months for non-resected patients.^31)^ Wong et al. reported that patients who underwent abdominal or pelvic tumor resection had a median survival of 47 months compared with 16 months for non-resected patients.^32)^
The most effective chemotherapy regimens with curative intent are still debated, but most are based on regimens used to treat other small round cell sarcomas, combining anthracyclines, alkylating agents, and vinca alkaloids. Kushner et al. recommended the P6 protocol (cyclophosphamide 2100 mg/m^2^ (days 1 and 2), doxorubicin 75 mg/m^2^ (days 1 and 2), and vincristine 0.67 mg/m^2^ (day 1) for courses 1, 2, 3, and 6, and ifosfamide 1800 mg/m^2^ (days 1–5) and etoposide 100 mg/m^2^ (days 1–5) for courses 4, 5, and 7), which, when combined with surgical treatment, resulted in a favorable 5-year survival rate of 35%.^33)^
HIPEC has been reported to be effective in cases with good response after chemotherapy and without extraperitoneal metastasis.^31)^ In the Phase II clinical trial of HIPEC for DSRCT following cytoreductive surgery (CRS), reported by Hayes-Jordan et al. in 2018, improvements were observed in both the 3-year overall survival rate and the 3-year recurrence-free survival rate.^30)^ However, some reports have not demonstrated the efficacy of HIPEC after CRS.^34,35)^ Therefore, the benefits of HIPEC are still unclear and need to be evaluated in prospective trials.
Cases of intestinal DSRCT reported previously in the English literature to date are summarized in Table 2. The 6 patients, including this case, were 3 males and 3 females, and their ages at diagnosis ranged from 14 to 56 years. DSRCT has a poor prognosis and requires multimodal therapy, but 3 patients did not wish to undergo it. All 3 patients who did not wish to undergo multimodal therapy have died.
Current research is focused on developing targeted immunotherapy using monoclonal antibodies directed at DSRCT antigens.^36)^ Two DSRCT cell surface antigens have been identified: GD2, recognized by the antibody 3F8, and the immunomodulatory molecule B7H3, recognized by the antibody 8H9.^37,38)^ Other clinical trials are exploring experimental treatments such as the receptor tyrosine kinase (RTK) inhibitor Pazopanib, dopamine-like receptor 2 antagonist ONC201, intraperitoneal radioimmunotherapy with 131I-omburtamab, and HIPEC^.30,39–41)^ Lamhamedi-Cherradi et al. employed enzalutamide and androgen receptor-directed antisense oligonucleotides (AR-ASO) to inhibit DSRCT cell growth induced by 5α-dihydrotestosterone, significantly decreasing xenograft tumor burden and highlighting the efficacy of androgen-targeted therapies.^42)^
Currently, potential therapeutic targets for DSRCT are under development.^36)^
CONCLUSIONS
We experienced a very rare case of DSRCT. DSRCT is a fatal disease that primarily affects adolescent and young adult males. Currently, there is no proven treatment. More case reports are essential to improve the management of this disease.
DECLARATIONS
Funding
No funding was received for this study.
Authors’ contributions
All authors managed the perioperative course.
NT, MT, KT, and YY performed the operation.
NT contributed to the drafting and the manuscript.
MT, DU, KI, SM, SM, TY, KN, YY, MN, KT, TH, DT, HY, MT, and YA contributed to the revision of the manuscript critically for important intellectual content and have made final approval of the manuscript.
All authors have read and approved the manuscript, and they are responsible for the manuscript.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethical approval and consent to participate
This work does not require ethical considerations or approval.
Consent for publication
Consent was obtained from the patient to publish the clinical and imaging data in this report.
Competing interests
The authors declare that they have no competing interests.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chen G Zhang Q Xia D. Abdominopelvic desmoplastic small round cell tumor with metastasis: a case report and literature review. Medicine (Baltimore) 2024; 103: e 37664.38579065 10.1097/MD.0000000000037664 PMC 10994471 · doi ↗ · pubmed ↗
- 2Zhou J Li Q Luo B Primary desmoplastic small round cell tumor of the submandibular gland: a case report and literature review. Diagn Pathol 2022; 17: 6.34996495 10.1186/s 13000-021-01183-3PMC 8742402 · doi ↗ · pubmed ↗
- 3Wang LL Ji ZH Gao Y Clinicopathological features of desmoplastic small round cell tumors: clinical series and literature review. World J Surg Oncol 2021; 19: 193.34193155 10.1186/s 12957-021-02310-6PMC 8247134 · doi ↗ · pubmed ↗
- 4Slim S Zemni I Bouida A Intraabdominal and ganglionic desmoplastic small round cell tumor: a case series. J Med Case Rep 2021; 15: 500.34635162 10.1186/s 13256-021-03094-9PMC 8507229 · doi ↗ · pubmed ↗
- 5Ladanyi M Gerald W. Fusion of the EWS and WT 1 genes in the desmoplastic small round cell tumor. Cancer Res 1994; 54: 2837–40.8187063 · pubmed ↗
- 6Gerald WL Rosai J. Case 2. Desmoplastic small cell tumor with divergent differentiation. Pediatr Pathol 1989; 9: 177–83.2473463 10.3109/15513818909022347 · doi ↗ · pubmed ↗
- 7Gerald WL Miller HK Battifora H Intra-abdominal desmoplastic small round-cell tumor. Report of 19 cases of a distinctive type of high-grade polyphenotypic malignancy affecting young individuals. Am J Surg Pathol 1991; 15: 499–513.1709557 · pubmed ↗
- 8Magrath JW Kang HG Hartono A Desmoplastic small round cell tumor cancer stem cell-like cells resist chemotherapy but remain dependent on the EWSR 1-WT 1 oncoprotein. Front Cell Dev Biol 2022; 10: 1048709.36506091 10.3389/fcell.2022.1048709 PMC 9732033 · doi ↗ · pubmed ↗