From Common Symptom to Critical Diagnosis: Vertigo as a Clue to Thrombotic Thrombocytopenic Purpura
Aysche J Stern, Nicholas R Munoz, Reda Khan, Chibuike C Agwuegbo, Salma Yasin, Marie Pearson, Fatima Gauhar

TL;DR
A rare blood disorder was misdiagnosed as a common inner ear issue, highlighting the need to consider TTP in patients with unusual symptoms and lab results.
Contribution
This case highlights the importance of recognizing TTP in patients with atypical presentations and non-specific symptoms.
Findings
The patient initially presented with vertigo and normal hemoglobin but later developed hemolytic anemia and severe thrombocytopenia.
She was eventually diagnosed with TTP and treated with plasma exchange and rituximab.
The case underscores the need for early suspicion of TTP despite initial atypical symptoms.
Abstract
Thrombotic thrombocytopenic purpura is a hematologic disease with a high mortality rate that affects multiple organ systems. It is caused by a deficiency of a metalloprotease known as the ADAMTS-13 enzyme. Patients can present with fever, hemolytic anemia, thrombocytopenia, kidney injury, and neurological symptoms. Here we present a case of a female with TTP who was initially diagnosed with benign paroxysmal positional vertigo (BPPV). Her initial symptoms were vertigo, blurry vision, and gait imbalance, which were accompanied by elevated total bilirubin, acute kidney injury, thrombocytopenia, and normal hemoglobin. When the patient followed up in her primary care clinic, she was found to have worsening renal function, hemolytic anemia, and severe thrombocytopenia. She was subsequently diagnosed with TTP and referred to the emergency department (ED) where she received emergent treatment…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
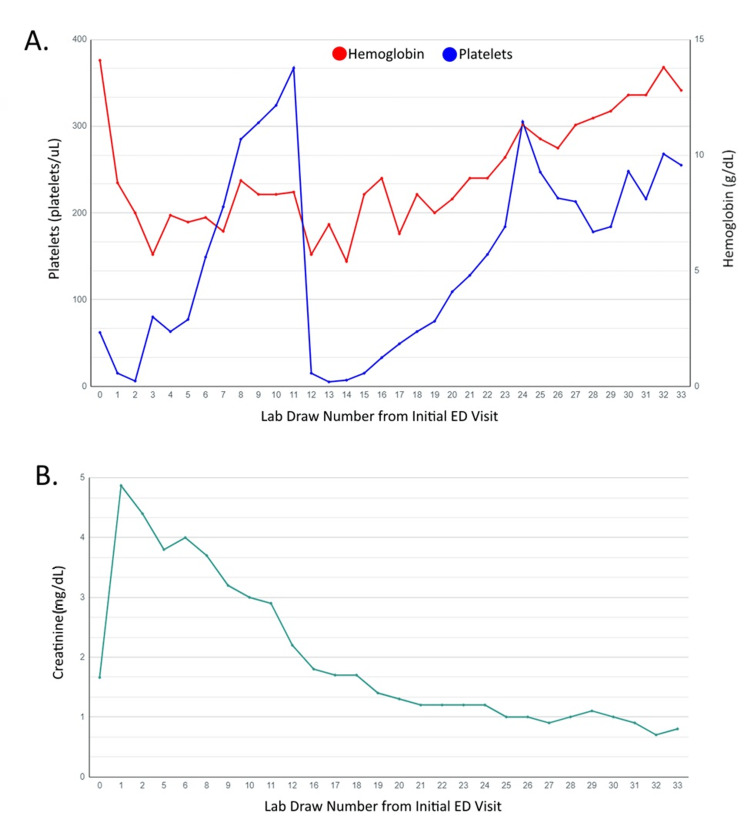 Figure 1
Figure 1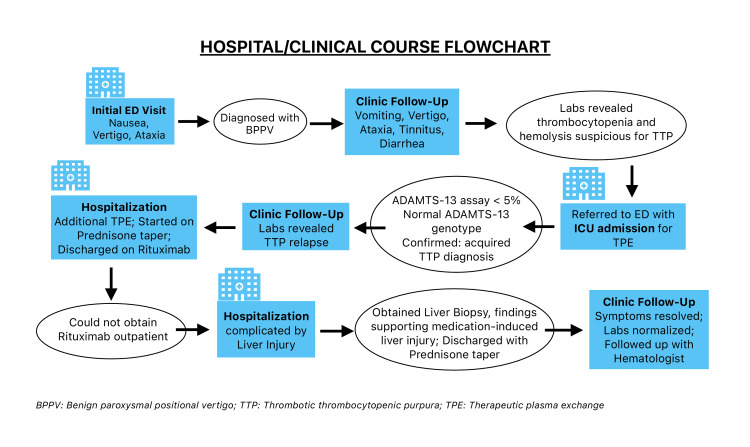 Figure 2
Figure 2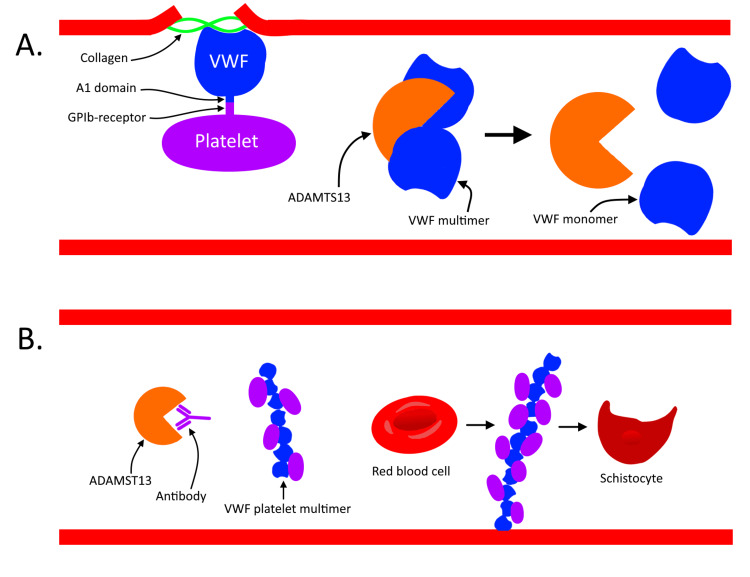 Figure 3
Figure 3| Test | Result | Normal Value |
| White Blood Cell Count | 7,600 cells/uL | 4,500-11,000 cells/uL |
| Hemoglobin | 7.4 g/dL | 12.0-16.0 g/dL |
| Hematocrit | 21.3% | 36-46% |
| Platelets | 6,000 platelets/uL | 150,000-400,000 platelets/uL |
| Blood Urea Nitrogen | 63 mg/dL | 8-20 mg/dL |
| Creatinine | 4.4 mg/dL | 0.6-1.2 mg/dL |
| Alkaline Phosphatase | 48 IU/L | 45-115 IU/L |
| Aspartate Aminotransferase | 36 IU/L | 8-40 IU/L |
| Alanine Transaminase | 21 IU/L | 8-40 IU/L |
| Reticulocyte Percentage | 4.10% | 0.5-1.5 % |
| Fibrinogen | 347 mg/dL | 150-350 mg/dL |
| Haptoglobin | <10 mg/dL | 50-150 mg/dL |
| Lactate Dehydrogenase | 1754 U/L | 40-90 U/L |
| Direct Bilirubin | 0.6 mg/dL | 0.0-0.3 mg/dL |
| Total Bilirubin | 1.9 mg/dL | 0.1-1.0 mg/dL |
| D-Dimer | 20.8 mg/L | <0.5 mg/L |
| Troponin | 65 ng/L | <14 ng/L |
| Homocysteine | 13.4 mmol/mL | 0.41-1.89 mmol/mL |
| Methylmalonic Acid | 0.22 mmol/mL | 150-370 mmol/mL |
| ADAMTS-13 | <5% | 40-140% |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComplement system in diseases · Mosquito-borne diseases and control · Hemoglobinopathies and Related Disorders
Introduction
Thrombotic thrombocytopenic purpura (TTP) is a rare and life-threatening blood disease caused by severe deficiency of a disintegrin and metalloprotease with thrombospondin type I repeats-13 (ADAMTS-13) enzyme, a von Willebrand factor (VWF)-cleaving protease [1]. This disease is characterized by microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and the formation of platelet-rich thrombi that ultimately result in end-organ ischemia primarily affecting the renal, neurological, and cardiac systems. Historically, a pentad of MAHA, thrombocytopenia, fever, and neurological and renal dysfunction was required for diagnosis of TTP. However, current guidelines consider unexplained MAHA and thrombocytopenia to be sufficient criteria for initiating treatment [2]. The case of a patient who was initially misdiagnosed with benign paroxysmal positional vertigo (BPPV) and later found to have TTP is presented.
Case presentation
Our patient is a female in her 40s with a history of hypertension, hypothyroidism, hyperlipidemia, and diabetes who initially presented to the emergency department (ED) with vertigo, nausea, and subjective gait imbalance. Initial labs showed a hemoglobin of 14.1 mg/dL, 62,000 platelets/uL, serum creatinine of 1.66 mg/dL, and total bilirubin of 2.6 mg/dL. Despite a normal physical exam, she was diagnosed with BPPV and discharged home. As her symptoms failed to improve, she presented to her primary care physician (PCP) three days later, endorsing vertigo, blurry vision, gait imbalance, nausea, tinnitus, and bilateral leg weakness. She further reported non-bloody diarrhea and vomiting for three days. The family history was negative for cancer, and she denied any smoking and illicit drug or alcohol use. The physical exam was normal other than a slow gait, without objective gait imbalance. A computerized tomography (CT) scan of the head showed no acute abnormalities but repeat laboratory work was significant for hemoglobin of 8.8 mg/dL, 15,000 platelets/uL, serum creatinine of 4.87 mg/dL, total bilirubin of 2.3 mg/dL, direct bilirubin of 0.6 mg/dL, and AST of 45 IU/L, as well as schistocytes on blood smear. Her PLASMIC score was calculated as six indicating a 72% risk for ADAMTS-13 deficiency. Together, these signs of worsening kidney injury with hemolytic anemia, thrombocytopenia, and neurological symptoms strongly suggested the diagnosis of TTP, and the patient was advised to immediately present to the ED.
After a presentation to the ED, she was admitted to the ICU, where follow-up laboratory analysis showed creatinine of 4.4 mg/dL, platelets of 6,000 platelets/uL, hemoglobin of 7.4 g/dL, negative indirect and direct antiglobulin test, total bilirubin of 1.9 mg/dL, direct bilirubin of 0.6 mg/dL, D-dimer of 20.8 ug/mL, elevated reticulocyte count of 3.6%, normal fibrinogen at 347 mg/dL, haptoglobin of <10 mg/dL, a normal troponin of 65 ng/L, homocysteine of 13.4 umol/L, methylmalonic acid of 0.22 mmol/mL, and lactate dehydrogenase of 1,754 U/L (Table 1). Her laboratory values indicated a past Epstein-Barr and cytomegalovirus infection. She tested positive for antinuclear antibody and Sjögren’s-syndrome-related antigen A autoantibodies. A hepatitis panel was negative. Stool studies were not completed as she did not have a bowel movement during hospitalization.
Due to high suspicion for TTP, treatment with therapeutic plasma exchange (TPE) with fresh frozen plasma transfusion in addition to prednisone 1 mg/kg daily was initiated despite pending results of the ADAMTS-13 activity testing. After five days of TPE, the patient's hemoglobin and platelet levels stabilized, and she was discharged with instructions to continue twice weekly complete blood counts until she could establish care with a hematologist. Her ADAMTS-13 activity assay resulted as <5%, with a normal ADAMTS-13 genotype confirming the diagnosis of acquired TTP.
At her outpatient follow-up after initial hospitalization, the patient reported normal bowel movements but continued to endorse balance issues and blurry vision. Laboratory values at this time resulted in a hemoglobin of 6.5 mg/dL, a platelet count of 11,000 platelets/uL, and a serum creatinine of 2.06 mg/dL, suggesting a relapse of TTP. She was hospitalized for eight days when she went into second remission. During this hospitalization, she received additional TPE and was started on prednisone 80 mg daily. After her platelets increased to greater than 11,000 platelets/uL, she was started on rituximab infusions and discharged.
However, after discharge, the patient was unable to receive rituximab on an outpatient basis and was ultimately hospitalized a third time for rituximab infusions. This hospitalization was complicated by liver injury with AST and ALT peaking at 927 U/L and 837 U/L respectively, and rituximab was subsequently discontinued. She underwent a liver biopsy showing sinusoidal dilation and edema with sparse portal inflammation without fibrosis or cirrhosis. Given those nonspecific findings, rituximab was assumed as cause of this iatrogenic liver injury.
Outpatient follow-up showed stable laboratory values including a normal hemoglobin level, platelet count, and creatinine, with no further relapse thereafter (Figure 1). The patient reported that her balance had improved; however, she continued to complain of dizziness and blurry vision. She denied symptoms suggestive of autoimmune disease such as dry eyes, dry mouth, rashes, joint pain, and muscle pain. Repeat ADAMTS-13 levels were normal two months after her initial ICU stay. Her liver function tests normalized, and she restarted her home medications metformin and rosuvastatin. Her symptoms resolved, allowing her to start exercising and she returned to work. She continues to follow up with her hematologist and PCP for regular check-ups (Figure 2).
Graph of the patient's platelet count (A), hemoglobin level (A), and creatinine level (B) starting from the initial visit to the emergency department. Blood draw 0: initial visit; blood draw 1: during follow-up with primary care; blood draw 2-11: intensive care unit admission; blood draw 12: post-hospital follow-up with primary care; blood draw 13-23: third hospitalization; blood draw 24-33: follow-up labs after the patient’s third hospitalization
Flowchart of the patient's clinical course. Figure courtesy of Salma Yasin.
Discussion
The classic pentad of symptoms in TTP is only present in 7% of patients [3]. Neurological symptoms are present in 40-80% of patients, 46% of patients have signs and symptoms of bleeding, and 30-40% of patients have gastrointestinal symptoms [3,4]. TTP has a median onset during the fourth decade of life. It is more common in females and African American patients [5].
Many of the typical symptoms found in TTP are caused by the embolization of platelet-rich thrombi to arterioles. During normal primary hemostasis, damage to the blood vessel wall causes bleeding and exposure of subendothelial collagen. VWF sequesters platelets to this exposed collagen and other proteins to achieve hemostasis. This interaction is mediated by the VWF A1 domain binding to the platelets GP1b receptor (Figure 3A) [6]. In TTP, deficiency of ADAMTS-13 causes accumulation of ultra-large von Willebrand factor (ULVWF) multimers and platelet aggregation, leading to the formation of thrombi and ultimately MAHA (Figure 3B) [2]. Causes are divided into two groups: acquired (>90% of cases) and congenital. In acquired disease, the development of autoantibodies is most commonly idiopathic (primary) but can also be associated with underlying disease processes (secondary) like bacterial infections and autoimmune diseases [1]. Other risk factors include pregnancy, human immunodeficiency virus infection, acute illnesses, or drugs [1]. In 10% of cases, autoimmune diseases later develop after a previous episode of TTP [1].
A. Normal physiology of disintegrin and metalloprotease with thrombospondin type 1 repeats-13 (ADAMTS-13), which cleaves von VWF multimers into monomers. During normal hemostasis, the VWF A1 domain binds to the platelet GP1b receptor, facilitating platelet adhesion. B. Pathophysiology of TTP. Antibodies targeting ADAMTS-13 block its enzymatic activity, leading to the accumulation of VWF-platelet multimers. These multimers cause red blood cell lysis and the formation of schistocytes. Additionally, the multimers may embolize to arterioles, causing ischemia in affected organs.TTP, thrombotic thrombocytopenic purpura; VWF, von Willebrand factor; GP1b, glycoprotein 1bFigure courtesy of Nicholas Munoz.
The appropriate workup for TTP includes a complete blood count with a smear, a complete metabolic panel, and an ADAMTS-13 level [3]. The PLASMIC score is a seven-component score used to risk stratify patients for the disease. An ADAMTS-13 level of <10% supports the diagnosis [3]. MAHA is diagnosed by schistocytes on blood smears and laboratory findings such as elevated lactate dehydrogenase, decreased haptoglobin, and elevated bilirubin [7].
Without treatment, TTP has a survival rate of about 10%. TPE increases the survival rate by 78% [8]. Treatment consists of TPE, rituximab, corticosteroids, and caplacizumab as this combination has been shown to reduce mortality [9]. TPE removes anti-ADAMTS-13 antibodies and replaces ADAMTS-13. Corticosteroids and rituximab suppress the immune response against ADAMTS-13, helping to maintain remission and reduce relapses [10]. The early use of rituximab with TPE has been shown to prolong the duration of remission. Caplacizumab is an anti-VWF antibody that prevents the interaction of VWF with the platelet glycoprotein Ib-IX-V receptor, decreasing microvascular thrombosis. Trials have shown that, compared to patients in the placebo group, patients who received caplacizumab had a shorter time to normalization of platelets, lower mortality and recurrence rates, and fewer major thromboembolic events [11]. For patients in remission with evidence of low plasma ADAMTS-13 activity, the use of rituximab has been shown to decrease relapses [12].
ADAMTS-13 activity <10% is highly predictive of relapse, maintaining activity above that level can prevent relapses [13]. If a patient's ADAMTS-13 activity is persistently <10%, rituximab therapy can prevent a relapse [14]. During remission, serial ADAMTS-13 levels should be monitored in addition to chemistry panels, complete blood counts, and LDH. After an acute episode, ADAMTS-13 levels can be measured monthly for three months, every three months for one year, then once or twice a year if the patient is stable [15].
At the time of her initial evaluation in the ED, our patient demonstrated some signs of TTP including vertigo, acute kidney injury, moderate thrombocytopenia, and elevated bilirubin levels. The diagnosis was likely missed because of normal hemoglobin levels. The subsequent laboratory values in the outpatient setting were more significant and, mainly due to obvious signs of hemolytic anemia, indicated TTP, which required immediate hospitalization. Further investigations revealed a positive antinuclear antibody and Sjögren’s-syndrome-related antigen A; however, she did not meet diagnostic criteria for either Sjögrens syndrome or lupus. While the cause of her TTP remains unclear, an underlying autoimmune disorder may have played a role.
Conclusions
TTP is a rare but life-threatening disease, and both prompt recognition and treatment are important to reduce the associated high mortality rate. This case is unique due to the absence of anemia despite other typical signs and symptoms like vertigo, acute kidney injury, thrombocytopenia, and elevated bilirubin levels at the initial presentation. TTP should be considered in patients with a variety of presentations and laboratory values suggestive of the disease, even in the presence of normal hemoglobin levels. TTP treatment relies on TPE, rituximab, caplacizumab, and corticosteroids. Relapse is common, and close outpatient monitoring is recommended.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Thrombotic thrombocytopenic purpura: pathophysiology, diagnosis, and management J Clin Med Sukumar S Lämmle B Cataland SR 5361020213354056910.3390/jcm 10030536 PMC 7867179 · doi ↗ · pubmed ↗
- 2Thrombotic thrombocytopenic purpura: 2012 American Society for Apheresis (ASFA) consensus conference on classification, diagnosis, management, and future research J Clin Apher Sarode R Bandarenko N Brecher ME Kiss JE Marques MB Szczepiorkowski ZM Winters JL 1481672920142413634210.1002/jca.21302 · doi ↗ · pubmed ↗
- 3Clinical and laboratory diagnosis of TTP: an integrated approach Hematology Am Soc Hematol Educ Program Chiasakul T Cuker A 530538201820183050435410.1182/asheducation-2018.1.530PMC 6246034 · doi ↗ · pubmed ↗
- 4Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015 Blood Adv Page EE Kremer Hovinga JA Terrell DR Vesely SK George JN 590600120172929670110.1182/bloodadvances.2017005124 PMC 5728353 · doi ↗ · pubmed ↗
- 5Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts 13 deficiency: comparison of incidence, demographic and clinical features Pediatr Blood Cancer Reese JA Muthurajah DS Kremer Hovinga JA Vesely SK Terrell DR George JN 167616826020132372937210.1002/pbc.24612 · doi ↗ · pubmed ↗
- 6The von Willebrand factor A 1-collagen III interaction is independent of conformation and type 2 von Willebrand disease phenotype J Mol Biol Machha VR Tischer A Moon-Tasson L Auton M 324742920172788947410.1016/j.jmb.2016.11.014PMC 5186406 · doi ↗ · pubmed ↗
- 7Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia Dis Markers Barcellini W Fattizzo B 635670201520152681949010.1155/2015/635670 PMC 4706896 · doi ↗ · pubmed ↗
- 8Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura N Engl J Med Rock GA Shumak KH Buskard NA 3933973251991206233010.1056/NEJM 199108083250604 · doi ↗ · pubmed ↗