A leucine responsive small RNA AbcR200 regulates expression of the lactate utilization (lut) operon in Acinetobacter baumannii DS002
Harshita Nagasai Yakkala, Ashok Kumar Madikonda, Sandhya Rani Behera, Vijaykumar Pillalamarri, Kashif Gulam Mohammad, Ganeshwari Dhurve, Prasad Tammineni, Suresh Babu Pakala, Dayananda Siddavattam

TL;DR
This paper identifies a small RNA, AbcR200, in Acinetobacter baumannii that regulates lactate utilization genes in response to leucine levels.
Contribution
The study reveals a novel leucine-responsive small RNA, AbcR200, that regulates lactate utilization through translational inhibition of lut mRNA.
Findings
AbcR200 is regulated by leucine via the Lrp transcription factor, with promoter activity peaking at 2 mM leucine.
The lut mRNA is a target of AbcR200, with sequence complementarity and elevated protein levels in abcR200 negative strains.
Lrp interacts with the AbcR200 promoter under low leucine conditions, but this interaction is disrupted in the presence of leucine.
Abstract
Noncoding small RNAs are essential for modulating bacterial gene expression, especially under carbon and nutrient-limited conditions. In this study, by using both in silico and molecular hybridization tools, we identified a carbon source responsive small RNA in Acinetobacter baumannii DS002. Expression of corresponding gene, abcR200, located at the intergenic region of omt (O-methyl transferase) and orf72 genes, is under the transcriptional control of a global transcriptional factor, leucine responsive regulatory protein (Lrp). A sequence motif that serves as a target for Lrp was found overlapping the abcR200 promoter (PabcR200). Chromatin immunoprecipitation demonstrated that Lrp oligomers, formed under low leucine conditions, strongly interacted to the PabcR200. However, the observed interactions were disrupted in the presence of leucine, as leucine promoted dissociation of Lrp to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
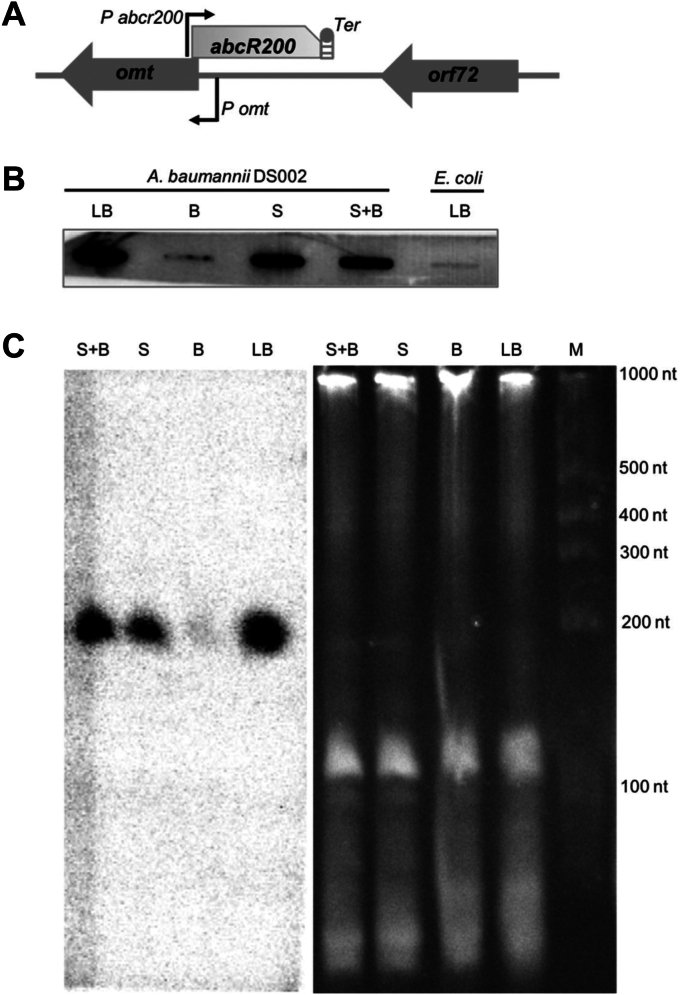 Figure 1
Figure 1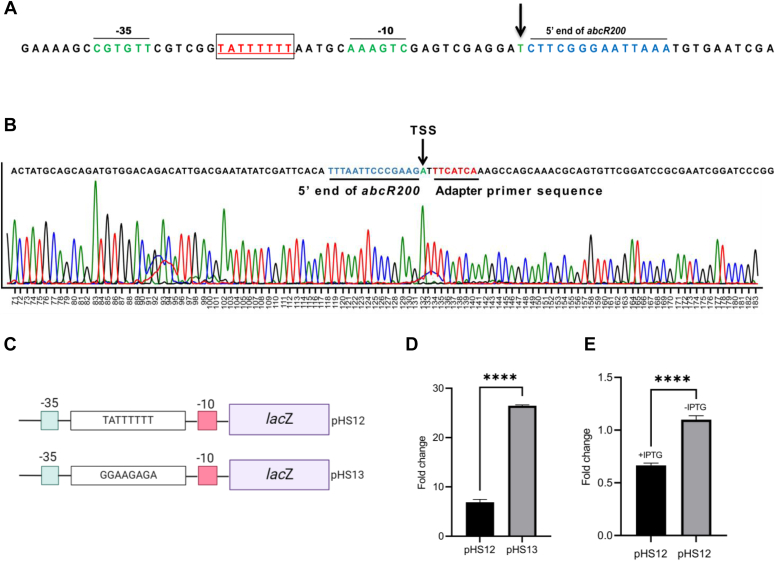 Figure 2
Figure 2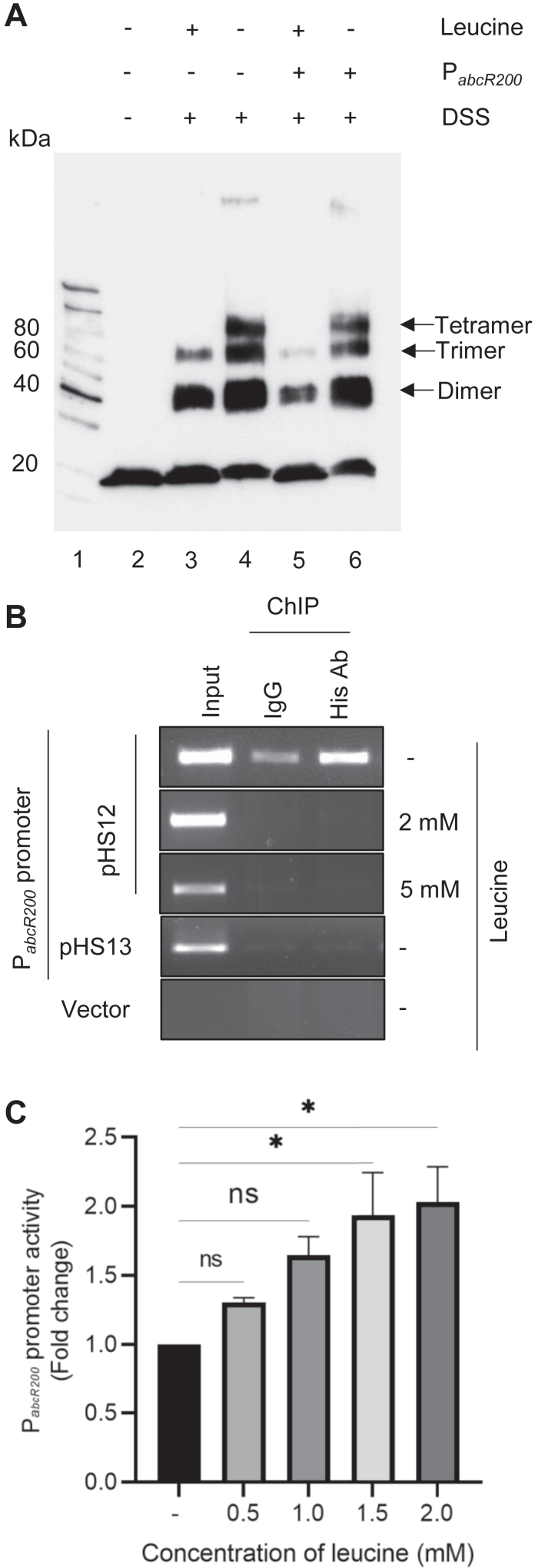 Figure 3
Figure 3 Figure 4
Figure 4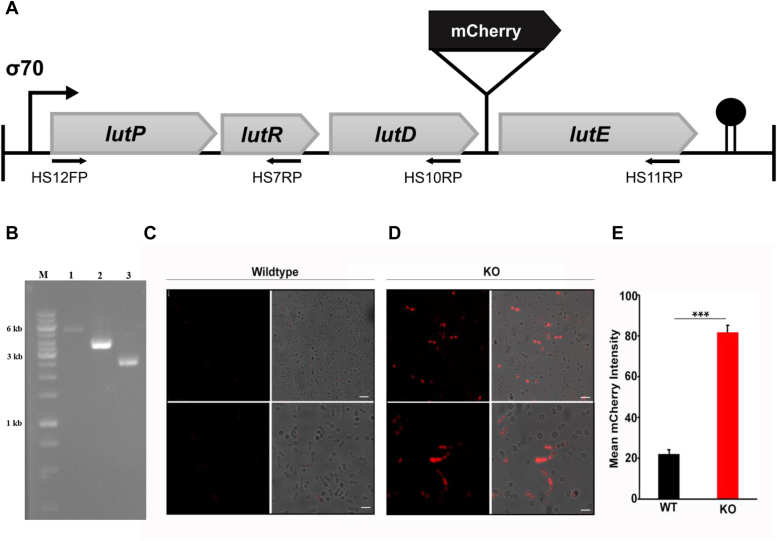 Figure 5
Figure 5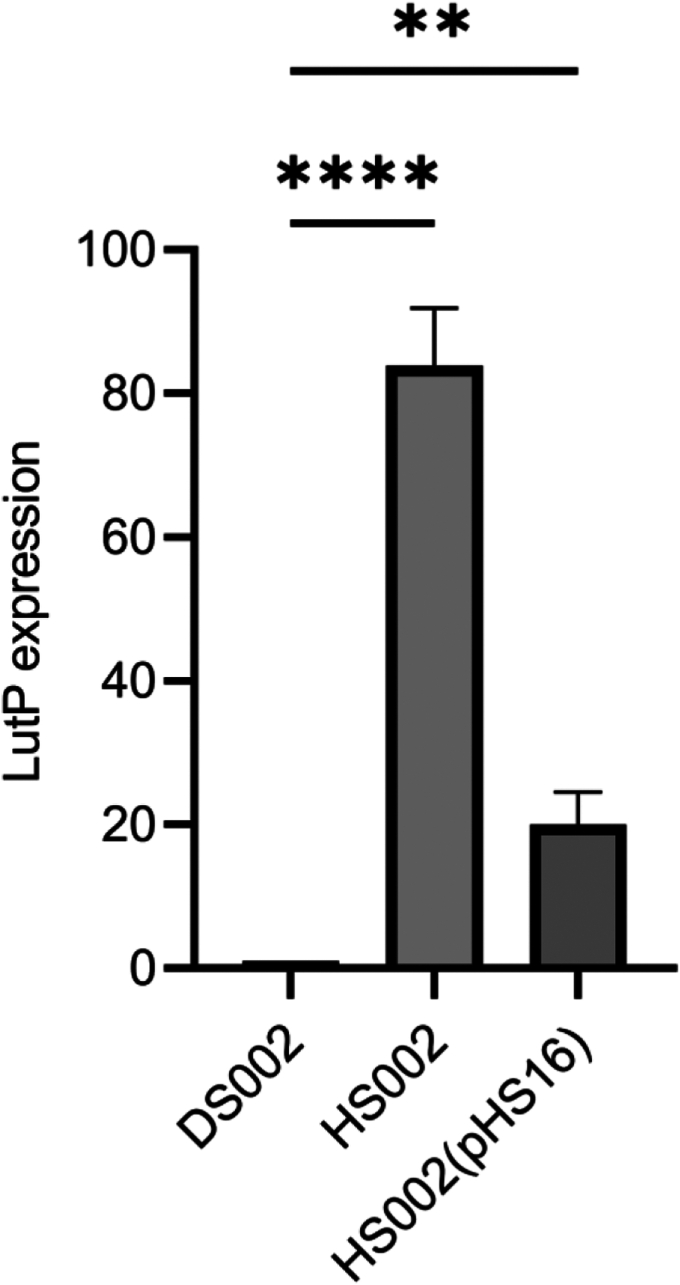 Figure 6
Figure 6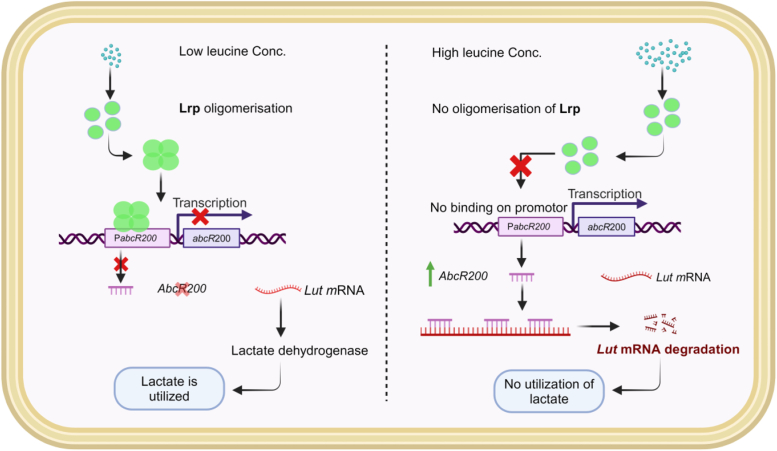 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Genetics and Biotechnology · Antibiotic Resistance in Bacteria · Genomics and Phylogenetic Studies
Genus Acinetobacter includes free-living saprophytic bacterial species found in soil, water, sewage, and foods. These ubiquitous organisms are also isolated as commensals from the skin of animals and humans, especially in staff working in hospitals and patients (1, 2, 3). Among them, Acinetobacter baumannii strains are opportunistic pathogens. They account for a significant portion of hospital-acquired infections caused by gram-negative bacteria (4). Before 1970s, A. baumannii strains were sensitive to most of the antimicrobials. Antibiotics like gentamicin, ampicillin, and nalidixic acid have effectively controlled A. baumannii infections (5, 6, 7). However, by late 1970s, they started showing resistance to most of the commonly used antibiotics. Soon, a pan drug-resistant strain of A. baumannii was isolated from hospital settings (8, 9, 10). A. baumannii strains exhibit a greater degree of genome plasticity (11, 12, 13). They acquire genetic material through conventional horizontal gene transfer or through membrane vesicles (14, 15, 16, 17, 18, 19). Intergenomic mobility of resistance genes is quite evident in certain strains of A. baumannii. The 89 kb resistance island identified in the genome of AYE strain contains forty-five drug-resistant genes. Most of them are orthologous to the resistance genes identified in Pseudomonas, Salmonella, and Escherichia coli (20).
The strains of A. baumannii are highly adaptable to a variety of ecological niches. They survive in soil, water, and on the surface of hospital ware as free-living organisms and quickly adapt to the pathogenic lifestyle (21, 22, 23). The ability to thrive on various carbon sources is assumed to be the reason behind the robust lifestyle of the strains (21). Most of the A. baumannii strains prefer succinate/acetate as a source of carbon. However, quite a few survive on disinfectants, recalcitrant xenobiotics, and phenolic compounds (24, 25, 26, 27).
Our laboratory isolated the A. baumannii strain DS002 from pesticide contaminated agricultural soils and sequenced its genome to gain better understanding on its biodegradation potential (23, 28). Similar to environmental strains of A. baumannii that exhibit a diauxic growth pattern (29), the DS002 genome contains genes encoding novel monooxygenases and dioxygenases, essential for initiating the degradation of several aromatic compounds (23). Notably, these genes are exclusively present in environmental isolates and are absent in clinical isolates (23). This selective enrichment of degradative genes, combined with distinct regulatory mechanisms, likely enhances the adaptive potential of environmental isolates of A. baumannii.
Bacterial small RNAs regulate a variety of biological processes through binding to their target mRNAs and regulatory proteins (30, 31, 32, 33, 34, 35). The regulatory roles of small RNAs are well studied in model organisms such as E. coli, Salmonella enterica, and, Staphylococcus aureus (36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49). However, despite its clinical and environmental relevance, similar studies are scarce in A. baumannii (50, 51). In this study, we analyzed the genome sequence of A. baumannii strain DS002, which has demonstrated the ability to use recalcitrant aromatic compounds as sole carbon source (23), to identify small RNAs involved in regulation of carbon metabolism. Our investigations uncovered a small RNA whose expression is regulated by the transcription factor, leucine responsive regulatory protein (Lrp). One of the key targets of this Lrp-regulated small regulatory RNA (sRNA) is the mRNA of the lactate utilization (lut) operon. Findings of this study highlight the intricate regulatory role of leucine responsive sRNA in regulating lactate metabolism in A. baumannii DS002.
Results
Carbon responsive small RNAs
Primary objective of this study was to identify small RNAs of A. baumannii DS002 involved in regulation of carbon catabolism. Initially, we have used in silico tools to identify small RNA coding sequences in A. baumannii DS002 genome (52). sRNA Identification protocol using high-throughput technologies (SIPHT) predicted existence of only 14 small RNA coding genes in the genome of A. baumannii DS002, whereas sRNA scanner predicted existence of as many as 266 small RNA coding genes. The predicted sRNA coding gene sequences (266 + 14) were then used as input to identify sequence motifs that serve as targets to the transcription factors involved in carbon catabolism. Interestingly, we predicted presence of consensus Lrp binding site overlapping the promoter region of four sRNA coding genes. Initially, we performed slot blot analysis to gain quick assessment on the expression pattern of these four small RNA coding genes (Figs. 1, A and B and S1). Total RNA isolated from the cells grown in nutrient rich (LB), preferred (succinate) and less preferred (benzoate) carbon sources were probed by the end labeled oligos complementary to the predicted sRNAs. Out of these four small RNA coding genes, only the one identified in the intergenic region of O-methyl transferase (omt) and orf72 genes (Fig. 1A) has shown clear carbon specific expression pattern (Fig. 1B). It was significantly upregulated in cultures grown in nutrient rich LB medium (Fig. 1B, lane LB) and completely repressed in cultures grown in minimal medium containing less preferred carbon source, benzoate (Fig. 1B, lane B). However, its expression was restored in cultures grown either in succinate or in cultures gown in the medium containing both succinate and benzoate (Fig. 1B, lanes S and S + B). This carbon responsive small RNA was given a four-letter code name, AbcR, (A. baumannii, “c” for carbon, “R” for responsive) followed by a number that indicates its predicted size. Since the predicted size of the small RNA is approximately 200 bases, it is designated as AbcR200. After gaining preliminary assessment on the expression pattern of abcR200, we have performed Northern blots to revalidate the predicted size and expression profile of abcR200 (Fig. 1C). Reconfirming the results obtained from slot blot analysis, the Northern blots have shown very low levels of abcR200 expression in cultures grown in the presence of benzoate (Fig. 1C, lane B). However, a prominent AbcR200 specific signal was detected in cultures grown in succinate (Fig. 1C, lane S) and succinate and benzoate (Fig. 1C, lane S + B), and its intensity has further increased in cultures grown in LB medium (Fig. 1C, lane LB). The size of the AbcR200 specific signal matched with the mobility of the marker RNA with a size of 200 nucleotides and it coincided with the predicted size of AbcR200.Figure 1**Expression of abcR200 in response to different carbon sources.**A, Organization of abcR200 gene in A. baumannii DS002. B, Slot blot analysis showing the expression of abcR200 in LB (LB), benzoate (B), succinate (S) and succinate and benzoate (S + B). The total RNA isolated from E. coli (lane E) served as negative control. C, Northern blot analysis showing the expression and size of AbcR200 in A. baumannii DS002 cells grown in succinate and benzoate (S + B), succinate (S), benzoate (B), and LB (LB).
Regulation of abcR200
As stated before, the abcR200 gene is identified in the intergenic region of omt and orf72 genes (Fig. 1A). The omt gene found upstream of abcR200 codes for O-methyl transferase, whereas the gene (orf72) identified downstream of abcR200 codes for a protein of an unknown function (Fig. 1A). In fact, the abcR200 has opposite transcription orientation when compared to the transcription of omt gene (Fig. 1A). The putative promoter motif of the abcR200 (P_abcR200) is identified at the 5′ end of the omt gene and hence the abcR200 overlaps with the 5′ region of omt gene (Fig. 1A). The functional status of predicted PabcR200_ was validated by performing both in vitro and in vivo studies. Initially, we have performed 5′ rapid amplification of complementary DNA (cDNA) ends (RACE) to determine transcription start site (TSS) of abcR200. The sequence of the 150 bp RACE product contained adaptor sequence followed by a sequence identical to the 5′ region of abcR200 gene (Fig. 2B). The sequence of abcR200 started with an adenine residue, indicating it as the TSS of abcR200 gene (Fig. 2, A and B). The promoter hexamers identified at −10 (CGGTAT) and −35 (TTGAAA) regions have shown similarities with the consensus σ^70^ dependent promoters (53). Furthermore, a sequence motif that showed significant identity with the consensus Lrp binding site (TATTTTTT) was identified overlapping the promoter motif of abcR200 gene (Fig. 2A). We have performed both genetic and biochemical assays to validate the functional status of Lrp binding site. Initially, we have constructed abcR200-lacZ fusions by including the WT (pHS12), and mutant (pHS13) Lrp binding sites (Fig. 2C) and then electroporated them into lrp positive E. coli AM001 cells. While growing lrp positive E. coli AM001 (pHS13) cells in nutrient rich LB medium, we observed a fourfold increase in β-galactosidase activity (Fig. 2D) compared to E. coli AM001 (pHS12) cells containing the abcR200-lacZ fusion with a WT lrp binding site (Fig. 2D). The repressive lrp positive genetic background and growth condition did not influence the promoter activity when mutations were introduced in the consensus Lrp binding site of abcR200 gene (Fig. 2D). These results were further validated by creating lrp negative genetic background in E. coli. The E. coli lrp negative (HS001) cells were then cotransformed with an expression plasmid, pHS11 coding A. baumannii DS002 Lrp protein (_Ab_Lrp^C6His^) from an IPTG inducible promoter along with an abcR200-lacZ fusion (pHS12) having WT Lrp binding site. The E. coli HS001 (pHS11+ pHS12) cells were then grown in nutrient rich LB medium both in the presence and absence of IPTG. Reconfirming the repressive role of Lrp, the abcR200 promoter was active only when the expression of _Ab_Lrp^C6His^ was not induced (Fig. 2E, lane (-IPTG)). However, there was a significant reduction in the activity of abcR200 promoter (Fig. 2E, lane (+IPTG)) when expression of _Ab_Lrp^C6His^ was induced by adding IPTG.Figure 2**Lrp regulates the expression of abcR200 gene.A, Sequence of the 5′ region of abcR200 gene. Transcription start site (TSS) is shown with inverted arrow. The −10 and −35 promoter hexamers are shown with green color font. Predicted Lrp-binding site shown in red color font is underlined and indicated in an open box. B, The sequence of the 5′ RACE product. The TSS is shown with an inverted arrow, the adapter (*green color* font) and the sequence of 5′ region of AbcR200 (red color font) is underlined. C, Model depicting the construction of abcR200 promoter lacZ fusions with WT (pHS12) and mutant (pHS13) Lrp binding sites. D, β-galactosidase activity levels in lrp positive AM001 cells carrying abcR200 promoter lacZ fusions, with WT (pHS12) and mutant (pHS13) Lrp binding sites. E, β-galactosidase activity levels in Escherichia coli HS001 (pHS11 + pHS12) grown in the presence and absence of IPTG. Data plotted are mean ± SD of three independent experiments. Statistical significance of data wherever applicable is indicated by ∗p < 0.05; ∗∗∗p < 0.001. Lrp, leucine responsive regulatory protein; RACE, rapid amplification of cDNA ends.
Lrp oligomers bind to PabcR200
Lrp exerts its influence over a number of bacterial genes by responding to intracellular leucine concentrations (54, 55, 56). When leucine concentration is low, Lrp adopts a high affinity DNA-binding oligomeric state. Conversely, in the presence of leucine, it dissociates into a transcriptionally inactive monomeric or dimeric state (54, 56). Since promoter assays indicated a potential role for _Ab_Lrp^C6His^ in regulating abcR200 gene expression (Fig. 2E), further studies were conducted to determine whether intracellular leucine concentration influences the regulation of abcR200. This proposed regulatory role of leucine was validated by performing both in vitro and in vivo experiments. We initially performed in vitro studies to investigate whether AbLRP^C6His^ oligomers form in the absence of leucine, and to assess if their formation is dependent on the abcR200 (PabcR200) promoter. The pure _Ab_Lrp^C6His^ (Fig. S2) incubated with a cross linker disuccinimidyl suberate (DSS), was resolved on SDS-PAGE (12.5%) and formation of _Ab_Lrp^C6His^ oligomers were detected by performing Western blots using anti-His antibodies (Fig. 3A). As expected, leucine promoted dissociation of _Ab_Lrp^C6His^ into dimeric and trimeric states (Fig. 3A lanes 3 & 5). The tetrameric _Ab_Lrp^C6His^ was only seen in reactions performed in the absence of leucine (Fig. 3A, lanes 4 & 6). Intriguingly, the DNA fragment containing the abcR200 promoter with the Lrp binding site had no effect on the formation of _Ab_Lrp^C6His^ tetramers, suggesting that the absence of leucine alone drives the formation of these tetramers (Fig. 3A lanes 4 & 6). After confirming the formation of AbLrp^C6His^ oligomers in the absence of leucine, we conducted chromatin immunoprecipitation (ChIP) analysis to determine whether these Lrp oligomers interact with the abcR200 promoter. The cell lysate prepared from a lrp negative E. coli HS001 strain containing an abcR200-lacZ fusion (pHS12) and a compatible expression plasmid (pHS11) coding AbLrp^C6His^ was immunoprecipitated using anti-His antibodies and the precipitate was then used to detect PabcR200. The ChIP assays convincingly demonstrated recruitment of AbLrp^C6His^ onto PabcR200 only in the E. coli cells grown in the absence of leucine (Figs. 3B and S3). Immunoprecipitates prepared from lysates of E. coli HS001 (pHS11 + pHS12) cells grown in the presence of exogenous leucine (2 mM and 5 mM) no such promoter recruitment was observed (Figs. 3B and S3). Revealing the specificity of AbLrp^C6His^ and PabcR200 interactions, no PabcR200 promoter was detected in immunoprecipitates performed using lysates of E. coli cells (pMMB206 +pHS11) containing expression vector and abcR200-lacZ fusion (Fig. 3B), or in cells having expression plasmid (pHS11) coding AbLrp^C6His^ and abcR-lacZ fusion (pHS13) with mutant lrp binding site (Figs. 3B and S3). The results of the ChIP assays, when considered alongside in vitro studies suggesting the formation of Lrp oligomers in the absence of leucine (Fig. 3A), strongly indicate that Lrp, in its oligomeric state, interacts with the Lrp-binding motif located within the promoter region of the abcR200 gene (Fig. 3B). Finally, we have performed promoter assays in E. coli HS001 (pHS12) cells to assess the influence of exogenous leucine on the expression of abcR200 gene. The E. coli cells (pHS12) were grown in the presence of different concentrations of exogenous leucine, and promoter assays were performed by measuring β-galactosidase activity (Fig. 3C). The promoter activity, as determined by measuring β-galactosidase activity, increased with an increase in exogenous leucine concentration and showed maximum activity at 2 mM (Fig. 3C). These results, together with the results of ChIP assays, clearly demonstrate the role of Lrp in regulating the expression of abcR200 gene by modulating its oligomeric state in response to exogenous leucine concentration.Figure 3PabcR200 andAbLrp^C6His^**interactions.**A, Leucine induced conformational changes in _Ab_Lrp^C6His^. Pure _Ab_Lrp^C6His^ (lane 2) incubated in the presence (lane 3) and absence (lane 4) of leucine was cross-linked using DSS (disuccinimidyl suberate) and separated on 12.5% SDS-PAGE, and the Western blot analysis was performed by probing with anti-His antibodies. The _Ab_Lrp^C6His^ dimers, trimers, and tetramers formed are shown with arrows. Formation of _Ab_Lrp^C6His^ tetramers is seen both in the absence (lane 4) and presence of PabcR200 (lane 6). Lane 1 represents the protein marker. B, The ChIP assays: The Escherichia coli HS001 (pHS11 + pHS12) grown in the absence and presence of leucine (2 mM and 5 mM) were induced to express _AB_LRP^C6His^. The clear lysate prepared from these cells were immunoprecipitated (IP) using anti-His antibodies, and the obtained precipitates were used to amplify PabcR200. PabcR200 amplification was only seen in cells grown in the absence of leucine, and no such amplification was observed in cells grown in the presence of leucine. Cell lysates prepared E. coli HS001 (pHS11 + pHS13), E. coli HS001 (pHS11 + pMP220) served as negative controls. C, Promoter assays were performed by measuring β-galactosidase activity levels in cells of E. coli HS001 (pHS11 + pHS12) cells grown in the absence and presence of 0. 5, 1.0, 1.5, and 2.0 mM leucine. Promoter activity increased with the increase of leucine concentration, and maximum activity was observed in cells grown in media containing 2.0 mM leucine. One-way Anova was used to plot the data and it represents mean ± SD of three independent experiments. Statistical significance of data wherever applicable is indicated by ∗p < 0.05; ∗∗∗p < 0.001. Lrp, leucine responsive regulatory protein; ChIP, chromatin immunoprecipitation.
Lactate utilization (lut) operon is part of abcR200 regulon
Bacterial sRNAs modulate expression of a multitude of genes by exerting their influence through the sequestration of regulatory proteins or direct interaction with target mRNAs (49). In this study, we report existence of a Lrp regulated small RNA coding gene, abcR200, in A. baumannii DS002. Lrp, is widely regarded as feast/famine regulatory protein and directly controls expression of significant number of genes that perform wide variety of functions in Gram negative bacteria (56). Intriguingly, in A. baumannii, Lrp is regulating the expression of a regulatory small RNA, AbcR200, and therefore expected to exert indirect influence on A. baumannii gene expression. We have performed additional experiments to unravel this regulatory interplay by identifying the mRNA targets of AbcR200. Initially we have generated abcR200 negative genetic background in A. baumannii DS002 and the abcR200 negative strain, HS002 was then used to determine the transcription profile by RNA sequence analysis (RNA sequence accession number, SRA: SAMN32537938; ID: 26171994). The generated transcription profile was then compared with the transcriptome of WT strain DS002 grown under identical growth condition. Approximately, 12 to 24 million raw reads were generated independently for the total RNA isolated from three biological replicates of WT and abcR200 negative A. baumannii HS002 strain (Table S1). These raw reads were then used to identify the list of differentially expressed genes by setting the cutoff value of p.adj <0.5 and log2 fold change greater than 1. Analysis of the sequence data following the set rigorous parameters led to the identification of 123 differentially expressed genes within the abcR200 null background, of which 66 and 57 genes were downregulated and upregulated, respectively (Tables S2 and S3). As shown in logarithmic plot, genes involved in lut were found in the list of significantly upregulated genes (Fig. S4 and Table S3). Lactate permease (lutP), transcriptional regulator (lutR), D-lactate dehydrogenase (lutE), and α-hydroxy-acid oxidizing enzyme (lutD) are all upregulated in abcR200 negative background, suggesting a repressive role of abcR200 on expression of lut genes.
The lut mRNA is complementary to AbcR200
As described in methods section, deletion of abcR200 invariably disrupts the omt gene. Consistent of the genetic deletion, the omt gene was also found in the list of downregulated genes in A. baumannii HS002 cells (Fig. S4 and Table S2). Therefore, the observed expression profile, especially the upregulation of lut gene expression was validated by performing additional experiments. As small RNAs influence gene expression by directly interacting with target mRNAs, we initially performed in silico studies to predict sequence complementarity between lut mRNA and AbcR200. These in silico predictions were then validated by quantifying lut mRNA in omt positive and abcR200 negative genetic background.
Generally, extensive sequence complementarity exists between cis-coded small RNAs and their target mRNAs (31). The trans-encoded small RNAs exert regulatory effect by establishing short imperfect base pairing with the target gene mRNAs (31). The AbcR200 is a trans-coded small RNA to lut operon. There exists 1.1 Mb distance between lut operon and abcR200 gene (23). Surprisingly, extensive sequence complementarity was noticed between various regions of lut mRNA and AbcR200 (Fig. 4A, rows I-III). Notably, a 134 bases long region having potential to form imperfect base pairing was found between AbcR200 and the 5′UTR of lutP. The sequence complementarity started from −119 region and it continued till +15, where “A” of start codon ATG is taken as +1 (Fig. 4A, row I). Due to existence of intra strand complementarity, especially between 5′ and 3′ regions, the AbcR200 sequence acquires an overall hair-pin loop structure with several stems and loops (Fig. 4A). The sequence of AbcR200 (43–182) that shows complementarity to lutP mRNA exists at the arm-II of the stem structure (Fig. 4A, row I). Similar interacting regions were found between AbcR200 and lut mRNA regions coding LutE, and LutR. However, they were found either near TSS of lutE or at the 3′ end of the coding region of LutR (Fig. 4A, rows II & III). The sequence found at the arm I (3–60) of AbcR200 showed complementarity with these regions. The length of interacting regions and predicted binding energies was determined following online tool IntaRNA 2.0, and was sufficiently strong to exert translational inhibition on lut mRNA (57).Figure 4Base pairing regions between andlut mRNA**.**A, Indicates predicted complementary regions between AbcR200 (black font) and lut mRNA (red font) corresponding to lutP (panel A, row-I), lutR (panel A, row-II), and lutE (panel A, row-III). Both negative and positive numbering is given to the lut mRNA sequences complementary to AbcR200. The “A” of start codon ATG is numbered with +1. The sequence upstream of it is shown with negative numbering, whereas downstream sequence is given positive numbering. B, SDS-PAGE and corresponding Western blots indicate ectopically expressed Lut proteins from an IPTG inducible promoter. The WT A. baumannii DS002 and abcR200 negative HS002 cells were transformed with expression plasmids pHS8, pHS7, and pHS6 coding LutP^C6His^, LutR^C6His^, and LutE^C6His^, respectively and their expression was monitored by performing Western blot analysis using anti-His antibodies. Lane 1 indicates molecular size markers, lanes + and – represent total proteins isolated from the WT DS002 and abcR200 negative HS002 cells of A. baumannii carrying plasmid pHS8 (row-I), pHS7 (row-II), and pHS6 (row-III). C, The omt positive and abcR200 negative genetic background was generated by transforming plasmid pHS14, coding O-methyl transferase (Omt) from an IPTG inducible promoter into A. baumannii HS002 cells. The total RNA isolated from A. baumannii HS002 (pHS14) and WT A. baumannii DS002 cells was used for the quantification of lut mRNA by performing qPCR. Graph represents concentration of LutP (row-I), LutR (row-II), and LutE (row-III) specific mRNAs in abcR200 positive (lane I) and negative (lane II) backgrounds. LutP, lactate permease; qPCR, quantitative PCR.
AbcR200 inhibits translation of lut mRNA
These in silico predictions were further validated by monitoring the expression of lut genes both in WT (DS002) and in HS002 cells of A. baumannii. Initially, we have constructed broad host range mobilizable expression plasmids coding LutR^C6His^ (pHS6), LutE^C6His^ (pHS7), and LutP^C6His^ (pHS8) by including the predicted interacting regions. Construction of such expression plasmids was possible as sufficient length of predicted interacting regions with AbcR200 was found down stream of translational initiation codon ATG (Fig. 4A, rows I, II, and III). These expression plasmids were then independently mobilized into WT (DS002) and abcR200 negative strain (HS002) of A. baumannii. The expression levels of these ectopically expressed proteins were monitored by probing with anti-His antibody. As expected, signals that matched with the predicted size of LutP, LutE, and LutR were detected in both WT and abcR200 negative cells (Fig. 4B, rows I, II, and III). However, the expression levels of LutP, LutE, and LutR were markedly lower in WT cells compared to their expression levels in HS002 cells, implying a negative impact of AbcR200 on the expression of these genes (Fig. 4B, rows I, II, and III, lanes indicated with negative ‘−’ sign). The faint signals observed in WT cells may be attributed to the induction of ectopically expressed lut genes, which were cloned under the control of a strong inducible promoter (Fig. 4B, rows I, II, and III, lanes indicated with positive “+” sign). Conversely, in HS002 cells, the absence of AbcR200 mediated translational inhibition resulted in significantly elevated expression levels of LutP, LutE, and LutR (Fig. 4B, rows I, II, and III). Multiple signals were noticed in HS002 cells expressing LutP, probably due to degradation of membrane associated LutP in cells grown in physiologically unfavorable condition for LutP expression (Fig. 4B, row I). The elevated expressions of proteins coded by lut genes in HS002 cells clearly support in silico predictions which suggest possible AbcR200 mediated translational inhibition of lut mRNA.
The aforementioned study refers to the expression of lut specific ORFs from an inducible promoter. In A. baumannii DS002 cells, the 5.4 kb lut cluster encompassing lutP, lutR, lutD, and lutE genes showed an operon-like organization (23). A consensus σ^70^ promoter is predicted upstream of the lutP gene and a putative Rho-dependent transcription terminator like motif is only seen downstream of the lutE gene (Fig. 5A). Absence of promoter and terminator motifs flanking lutR and lutD ORFs favors organization of lut gene cluster as one transcriptional unit. These in silico predictions were further validated by measuring the size of lut mRNA by performing RT-PCR. The lut mRNA was reverse transcribed by using a fixed forward primer specific to lutP gene, and reverse primers were designed taking the coding regions of lutR, lutD, and lutE genes. In agreement with the in silico predictions, which indicated the organization of the lut cluster as a single transcriptional unit, we successfully obtained an amplicon of approximately 5.4 kb in a RT-PCR reaction performed using forward and reverse primers, specific to lutP and lutE regions (Fig. 5B, lane 1).Figure 5**Confocal Studies.**A, Organization of lut operon in A. baumannii DS002. Putative promoter and terminator motifs of lut operon are shown with bent arrow and solid circle, respectively. Black solid arrow indicates ORF coding mChery inserted at the intergenic region of lutD and lutE. Solid lines below ORFs indicate sequence regions used while designing primers for performing RT-PCR. B, Agarose gel (0.8%) showing the size of amplicons generated in RT-PCR reactions. Lane M indicates molecular size markers, lanes 1 to 3 show amplicons obtained in an RT-PCR mix containing HS12FP as fixed forward and HS11RP (lane 1), HS10RP (lane 2), and HS7RP (lane 3) as reverse primers. C and D, Confocal images were obtained for abcR200 positive DS002 (pPS5) and negative HS002 (pPS5) cells of A. baumannii, respectively. Representative images were picked from three biological replicates. Each experiment had roughly 1000 cells for quantification. To be precise, the number of WT cells are 3760 and KO cells are 3047. E, Graph representing the mean fluorescence intensity obtained for abcR200 positive DS002 (pPS5) and negative HS002 (pPS5) A. baumannii cells. Intensity values plotted come from the average, and error bars represent the standard deviation. Student t test was done for p-values.
Given the polycistronic nature of lut mRNA, we have designed further studies to enhance our comprehension of the impact of AbcR200 on the translation of lut mRNA. As described in the methods sections, we have constructed a broad host range plasmid (pPS5) that carries entire lut operon of A. baumannii DS002 along with an additional ORF coding mCherry at the intergenic regions of lutD and lutE genes (Fig. 5A). The plasmid, pPS5 borne lut operon variant, lut’ codes mCherry, in addition to lut specific proteins from its native promoter identified upstream of lutP gene (Fig. 5A). The WT DS002 and HS002 strains of A. baumannii carrying plasmid, pPS5 were then used to monitor the expression of lut operon by measuring the fluorescence. Supporting the AbcR200 dependent translation inhibition of lut operon, the WT DS002 (pPS5) strain emitted less amount of fluorescence (Fig. 5, C and E) when compared to the amount of fluorescence emitted by the HS002 (pPS5) cells (Fig. 5, D and E). These findings align with in silico studies and increased expression of lut genes in HS002 cells of A. baumannii, strongly suggests inhibitory role of AbcR200 on the translation of lut mRNA (Fig. 4, A and B, rows I, II, and III).
As deletion of abcR200 disrupts omt gene, we have created abcR200 negative and omt positive genetic background in HS002 by transforming _Ab_Omt^C6His^ coding plasmid, pHS14 and quantified lut specific mRNA in the presence of ectopically expressed Omt (Fig. S5). Aligning with the whole body of the data generated in this study, even in the presence of ectopically expressed Omt protein, all lut genes were found upregulated in A. baumannii HS002 (pHS14) cells (Fig. 4C rows I, II, and III). Especially, the lutP gene expression has gone up by 35 folds in HS002 (pHS14) cells (Fig. 4C, row I). Finally, we have created abcR200 positive genetic background in HS002 cells by electroporating a broad host range plasmid, pHS16 generated by cloning the abcR200 gene along with its native promoter. The HS002 (pHS16) strain was then used to quantify the lutP specific mRNA. The expression levels of lutP-specific transcript got significantly reduced when we created abcR200 positive background in HS002 cells (Fig. 6). This genetic evidence provides conclusive evidence on AbcR200 dependent translational inhibition of lut operon in A. baumannii HS002 cells.Figure 6The abcR200 negative A. baumannii HS002 cells were electroporated with a broad host range plasmid, pHS16 carrying abcR200 gene. The total RNA isolated from HS002 (pHS16), HS002, and WT A. baumannii DS002 cells was used to quantify LutP mRNA by performing qPCR. Graph represents concentration of LutP mRNA in DS002, HS002, and HS002 (pHS16) cells. One-way Anova was used to plot the data, and it represents mean ± SD of three independent experiments. Statistical significance of data wherever applicable is indicated by ∗p < 0.05; ∗∗∗p < 0.001. qPCR, quantitative PCR
Discussion
Our investigations have identified Lrp-regulated small RNA, AbcR200 in A. baumannii DS002. Lrp, is a global transcriptional regulator and regulates nearly 40 genes in E. coli (58). This dual transcriptional regulator positively regulates genes involved in amino acid biosynthesis, transport, and negatively regulates amino acid degradation (59). In addition to the genes involved in amino acid metabolism, genes coding outer membrane porins, synthesis of pili, aminoacyl tRNA syntheses are also found as part of Lrp regulon in E. coli (59). Besides influencing the expression of various genes, the 18 kDa Lrp protein also plays a key role in maintaining the compact structure of the E. coli chromosome and regulates its own expression based on the cells nutritional status (59). In nutrient-rich medium, Lrp is made at minimal concentrations, whereas its expression goes up in cells grown in less preferred carbon sources (59). Coinciding with the published results, _Ab_Lrp^C6His^ oligomerized in the absence of leucine and interacted with lrp-binding motif identified overlapping abcR200 promoter. Since abcR200 is part of Lrp regulon, nutritional status of the culture medium has shown significant influence on expression of abcR200. Its expression was significantly high in LB grown cultures and showed moderate expression when cells are grown in succinate. Its expression was insignificant in cultures grown using benzoate, probably due to low levels of intracellular leucine concentrations that favors oligomerization of Lrp, the molecular state that makes Lrp as an active repressor. The cross-linking studies done both in presence and absence of leucine clearly supported the proposed hypothesis.
The present study suggests existence of leucine dependent signaling mechanism in sensing carbon status. A. baumannii DS002 grows in a variety of ecological niches and frequently encounters fluctuations in carbon source. The cell needs to quickly reorient expression pattern of its catabolic repertoire to facilitate usage of available carbon sources. Under carbon limiting conditions the intracellular amino acid pool serves as a quick source of carbon (60). Particularly, leucine catabolism is favored by the cell as it yields acetyl CoA and acetoacetate that feed directly into tricarboxy (tricarboxylic acidlic acid cycle. Depletion of intracellular leucine concentration, due to its usage as carbon source, has obvious consequences. It serves as a direct chemical signal to indicate carbon starvation and promotes conversion of otherwise inactive Lrp into an active repressor by promoting its oligomerization. The oligomeric Lrp represses expression of AbcR200, which has a direct role in translation inhibition of lut mRNA. This novel link revolving around leucine, Lrp, and AbcR200 regulate lactate catabolism in A. baumannii DS002 (Fig. 7).Figure 7Model depicting the interplay between abcR200 expression and regulation of lut operon expression in A. baumannii DS002. Low intracellular leucine concentration promotes Lrp oligomerization, a favorable molecular conformation required to repress abcR200 gene by interacting with P_abcR200. In the absence of AbcR200, Lut mRNA is translated to produce lactate dehydrogenase (LDH), enabling A. baumanni DS002 to use lactate as source of carbon and energy. Dissociation of Lrp into monomers and dimers in the presence of high intracellular leucine promotes detachment of Lrp from PabcR200_ and promotes derepression of abcR200 gene. Presence of AbcR200 prevents translation of lut mRNA and promotes its degradation. In the absence of lut mRNA translation, LDH is not synthesized, the enzyme required to promote utilization of lactate as source of carbon and energy. Lrp, leucine responsive regulatory protein.
Lactate is used by many pathogenic bacteria as sole source of carbon (61). Ability to use lactate as carbon source directly contributes for enhanced pathogenesis in bacteria like Neisseria gonorrhoeae, Neisseria meningitides, S. aureus, and Haemophilus influenzae (62). Pathogens prefer lactate over other sugars as it provides instantaneous energy (63). In a recent study it has been shown that the A. baumannii growth increased with parallel increase of lactate concentration in blood plasma (64). The transcriptome analysis done for A. baumannii cells isolated from bacteremia mice showed notable increase (7–8 folds) in expression of genes involved in lactate utilization (65). Induction of lut operons in A. baumannii cells isolated from bacteremia mice has clear link to this study. The abcR200 gene is conserved among all pathogenic A. baumannii strains including the strain isolated from bacteremia mice (Fig. S6). This study clearly established a role for abcR200 in regulation of lut operon. Unless AbcR200 expression is repressed, increase of lut-specific mRNA is not possible in cells isolated from infected mice. Further studies are required to understand the regulation of abcR200 expression during various stages of infection and to establish its role in pathogenesis. The current study presents experimental evidence linking intracellular leucine concentration and lactate catabolism and reveals yet another adaptive mechanism existing in A. baumannii DS002 to counter carbon limiting conditions effectively (Fig. 6).
Experimental procedures
Strains and media
Bacterial strains, plasmids, and primers used in this study are listed in main Tables 1 and 2, respectively. All biochemicals and enzymes used in DNA manipulations were procured from Thermo Fisher Scientific India Pvt Ltd, DSS Takara Bio India Pvt Ltd, and Merck India Pvt Ltd. The A. baumannii DS002 cells were grown at 30 °C either in LB medium or in minimal medium (275 mM K_2_HPO_4_, 8.8 mM KH_2_PO_4_, 12 mM NH_4_NO_3_, 2 mM MgSO_4_.7H_2_O, 50 μM Fe(SO_4_)3, and 0.1 mM CaNO_3_. 4H_2_O) containing succinate (5 mM) or benzoate (5 mM) or benzoate (2.5 mM) + succinate (2.5 mM) or lactate (5 mM) as a sole source of carbon. When required, antibiotics ampicillin (100 μg/ml), kanamycin (30 μg/ml), chloramphenicol (30 μg/ml), and gentamycin (20 μg/ml) were added to the growth medium. The antibiotic concentrations were reduced to half while growing the cultures in lactate containing minimal medium. The filter sterilized leucine (5 mM) was supplemented to the culture medium while assessing its influence on expression of lut operon. The E. coli strains were grown in LB medium at 37 °C. Routine gene manipulations, Western blots, nucleic acid hybridization techniques, and β-galactosidase assays were performed by following standard protocols (66, 67).Table 1. Strains and plasmidsS. No.Strain or plasmidGenotype or phenotypeReference or sourceStrains1Escherichia coli DH5αλsupE44, ΔlacU169 (Δ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi1 relA1(92)2E. coli Bl21- DE3F – ompT gal dcmlonhsdSB (rB –mB –) λ (DE3 [lacI lacUV5-T7p07 ind1 sam7 nin5]) [malB+]K-12 (λS)(93)3E. coli S17–1recAprohsdRRP42Tc::MuKm::Tn7 integrated into the chromosome.(94)4JW0872–2F-, Δ (araD-araB) 567, ΔlacZ4787 (::rrnB-3), λ -, Δlrp-787::kan, rph-1, Δ (rhaD-rhaB) 568, hsdR514(82)5E. coli HS001K-12 F– λ – ilvG– rfb-50 rph-1Δlac ΔlrpThis study6E. coli AM001K-12 F– λ – ilvG– rfb-50 rph-1, Δlac(95)8Acinetobacter baumannii DS002Native WT strain. Cm^r^, Sm^r^(28)9Acinetobacter baumannii HS002Acinetobacter baumannii DS002 derivative, generated by deleting abcR200gene.This studyPlasmids1pTZ57R/TAmp^r^, TA vector used for cloning of PCR products.Thermo Fisher Scientific, USA2pMD20Amp^r^, TA vector used for cloning of PCR products.Takara Bio, India3pMMB206Cm^r^, low copy, broad host range, mobilizable expression vector. Drives transcription of cloned genes from an IPTG inducible tac promoter.(96)4pYH206Amp^r^, derivative of pMMB206This study5pMP220Tet^r^, promoter probe vector.(78)7pET23bAmp^r^. The T7 promoter containing expression vector. Facilitates expression of cloned genes with C-terminal 6xHis-tag.Novagen8pJQ210Gm^r^, Suicidal vector used for generatingabcR200 knockouts in A. baumannii DS002.(83)9pUC4KIXXKm^r^, contains excisable kanamycin cassette.(97)10pHS1Gm^r^, pJQ210 derivative. Generated by ligating upstream and downstream flanking regions (500 bp) of abcR200as NotI fragment. The abcR200 is replaced with an unique BamHI site.This study11pHS2Gm^r^, Km^r^, and pHS1 derivative. Generated by inserting the kanamycin cassette at the unique BamHI site.This study12pHS3Amp^r^, expression plasmid generated by cloning lutR gene of A. baumannii DS002 in pET23b as NdeI and XhoI fragment. Codes LutR^C6His^.This study13pHS4Amp^r^, expression plasmid generated by cloning lutE gene of A. baumannii DS002 in pET23b as NdeI and XhoI fragment. Codes LutE^C6His^.This study14pHS5Amp^r^, expression plasmid generated by cloning lutP gene of A. baumannii DS002 in pET23b as NdeI and XhoI fragment. Codes LutP^C6His^.This study15pHS6Amp^r^, pHY206 derivative, generated by cloning lutR gene amplified from pHS3 as SmaI and SalI fragment. Codes LutR^C6His^.This study16pHS7Amp^r^, pHY206 derivative, generated by cloning lutE gene amplified from pHS4 as EcoRI and SalI fragment. Codes LutE^C6His^.This study17pHS8Amp^r^, pHY206 derivative, generated by cloning lutP gene amplified from pHS5 as EcoRI and SalI fragment. Codes LutP^C6His^.This study18pHS9Amp^r^, generated by cloning 5′ RACE productpTZ57R/T.This study19pHS10Amp^r^, Expression plasmid generated by cloning lrp gene of A. baumannii DS002 in pET23b as NdeI and XhoI fragment. Codes Lrp^C6His^.This study20pHS11Cm^r^, pMMB206 derivative, generated by cloning lrp gene amplified from pHS10 as EcoRI and HindIII fragment. Codes for Lrp^C6His^.This study21pHS12Tet^r^, generated by cloning promoter region of abcR200 as EcoRI-PstI fragment in pMP220.This study22pHS13Tet^r^, generated by cloning mutant promoter of abcR200 in pMP220 EcoRI-PstI fragment.This study23pPS1Amp^r^, generated by cloning lut operon of A. baumannii DS002 in pMD20.This study24pPS2Amp^r^, pPS1 derivative, generated by introducing XhoI and NheI sites in the intergenic region of lutD and lutE genes of lut operon.This study25pPS3Amp^r^, pPS2 derivative, generated by inserting ORF coding mCherry at the intergenic region of lutD and lutE genes of lut operon as XhoI and NheI fragment.This study26pTSR6KKm^r^, Shuttle vector constructed during indigenous plasmid of pTS4 of A. baumannii DS002(28)27pPS4Amp^r^, pTSR6K derivative, generated by replacing kanamycin cassette of pTSR6K with bla gene. The bla gene is inserted as XhoI and BamHI fragment.This study28pPS5Amp^r^, pPS4 derivative, generated by ligating lut operon obtained from pPS3 as BamHI and SalI fragment. Contains ORF coding mCherry at the intergenic region of lutD and lutE genes.This study29pHS15Amp^r^ generated by cloning omt gene as NdeI and XhoI fragment in pET23b. Codes for Omt^C6His^This study30pHS14Cm^r^ generated by cloning omt gene amplified from pHS15 in pMMB206 as EcoRI and HindIII fragment. Codes for Omt^CFLAG^This study31pHS16Amp^r^ generated by cloning abcR200 in pYH206This studyTable 2List of primers.S.No.NameSequence (5′ to 3′)Description1HS1 FPCGTCAATGTCTGTCCAC ATCTGCTGCATAGAbcR200 specific forward primer used while performing real time PCR to quantify the transcript of abcR200 gene2HS1 RPTCCCCTTATGCGCTTCA GTTTAAGGGGATTTGC TCAbcR200 specific reverse primer used while performing real time PCR to quantify the transcript of abcR200 gene3HS3GACAGGTGTAGACGAC GTA TCAGTTTAAATAbcR200 specific DNA oligonucleotide probe used while performing Northern blot analysis.4HS4 FPATCCTCTAGCGGCCGC GGTTTCACGAATGCCT TCAATAForward primer used to amplify abcR200 upstream region. The NotI site appended to facilitate cloning is shown in bold case.5HS4 RPATCGTCAGGATCCTCC CGAAGATCCTCGACTC GACReverse primer used to amplify abcR200 upstream region. The BamHI site appended to facilitate cloning is shown in bold case.6HS5 FPATCGCTTAGGATCCGA CGTATTGCGGGTTAAG TCAGGForward primer used to amplify abcR200 downstream region. The BamHI site appended to facilitate cloning is shown in bold case.7HS5 RPTACGTCATGTCGACGG TGGGTTGCGCCAGCTC TCCReverse primer used to amplify abcR200 downstream region. The SalI site appended to facilitate cloning is shown in bold case.8HS6 FPGCGTCAGCGTGTGCTG AATGACATGTPrimers used to quantify transcript of lutD gene by performing real time PCR.9HS6 RPCCACATCGGGCGATTA ATTGCAGGTGC10HS7 FPGTACTGATCAGTCGCC GTGGCGATGGPrimers used to quantify transcript of lutR gene by performing real time PCR.11HS7 RPCGTAACGCGGCATACC ATGCAGTC12HS8 FPACGCGTCAGTACCGTC AAGGTCGTCPrimers used to quantify transcript of lutE gene by performing real time PCR.13HS8 RPGACTTGCTTGCCGTCAT GGATGACTTG14HS9 FPGGCGATTATGGATGGC TGGCGTGGPrimers used to quantify transcript of lutP gene by performing real time PCR.15HS9 RPCAGAGTCTGACCCGCT TCAGGTTC16HS10 FPCTTAGTCATATGATGA GAATCTCCGATCAAGT GForward primer used to amplify lutR gene from A. baumannii DS002 chromosomal DNA. The NdeI site appended to facilitate cloning of lutR gene in pET23b vector is shown in bold case17HS10 RPGATAATCTCGAGTTTT GAATCAACTCTATTTA AACGGReverse primer used to amplify lutR gene from A. baumannii DS002 chromosomal DNA. The XhoI site appended to facilitate cloning of lutR gene in pET23b vector is shown in bold case.18HS11 FPCTATTGCATATGATGC AAACATTTTCTCCTCA AGCAGForward primer used to amplify lutE gene from A. baumannii DS002 chromosomal DNA. The NdeI site appended to facilitate cloning of lutE gene in pET23b vector is shown in bold case.19HS11 RPGACCAACTCGAGAGAT TTAGCCTTACCReverse primer used to amplify lutE gene from A. baumannii DS002 chromosomal DNA. The XhoI site appended to facilitate cloning of lutE gene in pET23b vector is shown in bold case.20HS12 FPGAGGTGCATATGATGC TCAATATGTGGCAACForward primer used to amplify lutP gene from A. baumannii DS002 chromosomal DNA. The NdeI site appended to facilitate cloning of lutP gene in pET23b vector is shown in bold case.21HS12 RPGACATTCTCGAGTGGA ATCATCCACGGAACCA GReverse primer used to amplify lutP gene from A. baumannii DS002 chromosomal DNA. The XhoI site appended to facilitate cloning of lutP gene in pET23b vector is shown in bold case.22HS13 FPCCTATGGATCCCATTCA AATATGTATCCGCTForward primer used to amplify bla gene from pGEX vector.23HS13 RPGTCTGACAGGGATCCA TGCTTAATCAReverse primer used to amplify bla gene from pGEX vector.24HS14 FPAACAGAATTCAGGGA GACCACAACGGTTTCC CForward primer used to amplify genes cloned in pET23b along with sequence coding C-terminus His-tag. The EcoRI site appended to facilitate cloning into pYH206 is shown in bold case.25HS15 FPAACACCCGGGAGGGA GACCACAACGGTTTCForward primer used to amplify genes cloned in pET23b along with sequence coding C-terminus His-tag. The SmaI site appended to facilitate cloning into pYH206 is shown in bold case.26HS15 RPCCTTGTCGACCTAGTT CCTTGTCGACCTAGTTReverse primer used to amplify genes cloned in pET23b along with sequence coding C-terminus His-tag. The SalI site appended to facilitate cloning into pYH206 is shown in bold case.27Adapter outer primerGCUGAUGGCGAUGAA UGAACACUGCGUUUGC UGGCUUUGAUGAAAAdapter specific forward primer used while performing 5′ RACE.28HS31 RPGTCATAAACGTTTAAA ATACGCCTATabcR200 specific outer reverse primer used while performing 5′RACE.29HS32 RPTTAAATGTGAATCGAT ATATTCGTCAAabcR200 specific inner reverse nested primer used while performing 5′RACE.30HS33 FPAGATAGAATTCAGCCA TTTCTGCATGAGTATCAForward primer used to amplify abcR200 promoter region. The EcoRI site appended to facilitate cloning is shown in bold case.31HS33 RPCATGACTGCAGGCAGC AGATGTGGACAGACAReverse primer used to amplify abcR200 promoter region. The PstI site appended to facilitate cloning is shown in bold case.32HS34 FPGCATAGGAATTCGATG ATTTGAAAAGCCGTGT TCGTCGGGGAAGAGAA ATGCAAAGTCGAGTForward primer used to amplify abcR200 promoter region with mutations in LRP binding motif. The EcoRI site appended to facilitatecloning is shown in bold case. The mutated sequence is shown in bold italics.33HS34 RPACGGATCTGCAGCTCA TTCTATAATCAACCAAReverse primer used to amplify abcR200 promoter region with mutations in LRP binding motif. The PstI site appended to facilitate cloning is shown in bold case.34HS35 FPGAAATCTACGTATGGC GTGGACAGACGForward primer used to amplify lrp gene from E. coli genome.35HS35 RPAAGGCGGCGGCCGCTA CTTAACTTTGReverse primer used to amplify lrp gene from E. coli genome.36HS36 FPCAGGTACATATGCGCC CCTTAGATCGTAForward primer used to amplify lrp gene from A. baumannii chromosomal DNA. The NdeI site appended to facilitate cloning of lrp gene in pET23b vector is shown in bold case.37HS36 RPGATGTACTCGAGCTTG CTCACATCGAGATATAReverse primer used to amplify lrp gene from A. baumannii chromosomal DNA. The XhoI site appended to facilitate cloning of lrp gene in pET23b vector is shown in bold case.38HS37 RPCCTTAAGCTTCTAGTT ATTGCTCAGCGGTGGCReverse primer used to amplify gene cloned in pET23b with C-terminus His-tag. The HindIII site appended to facilitate cloning into PMMB206 is shown in bold case.39PS1 FPCAAACG GGATCC CGGATATCATAAGCTT TAAAACForward primer used to amplify lut operon from A. baumannii genomic DNA.40PS1 RPCAAACG CTGCAG AAACCAAAAAGGTATG TTCAGReverse primer used to amplify lut operon from A. baumannii genomic DNA.41PS2 FPAACCTTTTCTCGAGCA AACCGTACTTCCTTGTA CGGForward primer used to create XhoI restriction site between lutD and lutE to introduce mCherry between lutD and lutE of lut operon42PS2 RPTACGGTTTGCTCGAGA AAAGGTTTTTGGTTACT TTTReverse primer used to create XhoI restriction site between lutD and lutE to introduce mCherry between lutD and lutE of lut operon43PS3 FPTGTACGGTTGCTAGCC AGGCGGCATCCATGCC GCCAForward primer used to create NheI restriction site between lutD and lutE to introduce mCherry between lutD and lutE of lut operon44PS3 RPTGCCGCCTGGCTAGCA ACCGTACAAGGAAGTA CGGTReverse primer used to create NheI restriction site between lutD and lutE to introduce mCherry between lutD and lutE of lut operon45PS4 FPATACCGCTCGAGCATT GTTTTATAAGTGAGAA ATGAForward primer used to amplify mCherry for inserting between lutD and lutE of lut operon46PS4 RPATACTAGCTAGCCTAC TTGTACAGCTCGTCCAT GCReverse primer used to amplify mCherry for inserting between lutD and lutE of lut operon47PS5 FPTATCACTCGAGCGCGG AACCCCTATTTGForward primer used to amplify bla gene from pGEX4T1. The XhoI site appended for replacing kanamycin cassette of pTSR6K is shown in bold.48PS5 RPGTGAGTGGATCCTTAC CAATGCTTAATCAGTReverse primer used to amplify bla gene from pGEX4T1. The BamHI site appended for replacing kanamycin cassette of pTSR6K is shown in bold.49MET FPCGCGAATTCAACATAC AGGTAAATTTGACTAT GCAGCForward primer used to amplify omt gene from the genomic DNA of A. baumannii DS00250MET RPTACTAAGCTTTTACTTGT CGTCATCGTCTTTGTAGTC TTTCACAATTGCAATTGReverse primer used to amplify omt gene from the genomic DNA of A. baumannii DS00251ChIP FPAGCCATTTCTGCATGA GTATCACGForward primer used to amplify PabcR200 promoter region of abcR200 from IPs (immunoprecipitates) obtained from the lysates of E. coli AM001 (pHS12) cells.52ChIP RPGCAGCAGATGTGGACA GACATTGAReverse primer used to amplify PabcR200 from IPs obtained from the lysates of E. coli AM001 (pHS12) cells of A.baumannii that binds to LRP53ChIP-LM- FPGCATAGGAATTCGATG ATTTGAAAAGCCGTGT TCGTCGGGGAAGAGAA TGCAAAGTCGAGTForward primer used to amplify PabcR200 having mutant Lrp binding site from IPs obtained from the lysates of E. coli AM001 (pHS13) cells.54ChIP-LM- RPACGGATCTGCAGCTCA TTCTATAATCAACCAAReverse primer used to amplify PabcR200 having mutant Lrp binding site from IPs obtained from the lysates of E. coli AM001 (pHS13) cells55AbcR-FPCCATGATTACGAATTC CACGTCCTAACCAGAT TGTACTGForward primers used to amplify abcR200 gene from genomic DNA of A.baumannii to facilitate infusion cloning56AbcR-RPTTGGCTGCAGGTCGAC TACTTGGGGACCTGAC TTAACCCGReverse primers used to amplify the abcR200 gene from genomic DNA of A.baumannii to facilitate in-fusion cloning57Infusion-FPGTCGACCTGCAGCCAA GCTTGGCACTGForward primers to amplify pYH206 for in- fusion cloning58Infusion-RPGAATTCGTAATCATGG TCATAGCTGTTTCCTGReverse primers used to amplify pYH206 for in-fusion cloning
In-silico tools
Prediction of small RNAs in A. baumannii DS002 genome was made using both sRNA identification protocol using high-throughput technologies (68) and sRNA scanner (69). The predicted small RNAs were then analyzed to predict prokaryotic promoter elements using Bprom (70). Sequence found 100 bp upstream and 30 bp downstream of the predicted sRNA promoters was then used as input to predict transcription factor binding sites using MEME suite (71). The MEME predicted signature sequences were further validated by TOMTOM to obtain similarity scores by comparing with the transcription factor binding motifs available in the data base (72). The sRNA/mRNA interactions were predicted using IntaRNA 2.0 (57). The potential promoter and terminator elements of sRNA coding genes and lut operon were predicted by using web-based tools Bprom (73) and Arnold (74), respectively, using default parameters. Online tool Mfold (75) was used to predict secondary structure of AbcR200.
Expression profiling of small RNAs
Slot blot analysis was done to assess expression profile of in silico predicted small RNAs in response to various carbon sources. Oligonucleotides complementary to the predicted sRNA sequences were designed and used as probe (Table 2). The probes were end-labeled following standard procedures, and slot blot experiments were performed as described elsewhere (76). Briefly, the cells were grown to mid-log phase either in LB or in minimal medium containing succinate or benzoate or both succinate and benzoate as sole source of carbon. Total RNA was isolated from these cultures following procedures described elsewhere (77). After assessing the integrity of isolated RNA, equal amounts of RNA (10 μg) were taken to make slots on nitrocellulose membrane cut to the size of the minifold (Bio-Rad Bio-Dot SF, Schleicher and Schuell Minifold II) and used to perform hybridization. While performing Northern blots, the isolated total RNA (40 μg) was separated on a 7M urea polyacrylamide gel (16%), and hybridization was performed following standard protocols using end-labeled oligo (5′-GACAGGTGTAGACGACGTATCAGTTTAAAT-3′) complementary to abcR200 (76).
Determination of TSS
Briefly, 5′ RACE experiments were performed to determine TSS of abcR200 gene. The 5′ RACE was performed using First Choice RLM-RACE Kit (Ambion Life technologies) following manufacturers protocol. RNA was isolated from A. baumannii DS002 cells grown to late log phase (absorbance of 1) in LB medium. After establishing RNA integrity, 5 μg of DNase-treated RNA was taken in sterile eppendorf tube and treated with tobacco acid pyrophosphatase to remove pyrophosphate from full-length mRNA molecules. Following removal of pyrophosphate, 45 mer RNA adapter oligonucleotide was ligated by incubating the tobacco acid pyrophosphatase-treated RNA at 37 °C for 1 h in presence of T4 RNA ligase. The adapter ligated RNA was then used as template for first strand cDNA synthesis. The cDNA thus synthesized was used to amplify the 5′ end of abcR200. A nested PCR was performed using adapter specific outer forward primer and gene specific reverse primers (HS31RP and HS32RP). The amplicon obtained was cloned in T-vector. The recombinant plasmid, pHS9 was used to determine sequence of RACE product and to establish transcription start point of abcR200.
Construction of abcR200-lacZ transcriptional fusions
The promoter fusions of abcR200 were generated by cloning the predicted promoter along with the Lrp-binding site in promoter test vector pMP220 (78). The abcR200 promoter region was amplified from genomic DNA of A. baumannii DS002 using primer set (HS33FP/HS33RP) appended with EcoRI and PstI restriction sites. The amplicon was gel extracted and digested with EcoRI and PstI before ligating it into pMP220 digested with similar enzymes. The resulting promoter fusion (abcR-lacZ) construct was named as pHS12. Promoter sequence with mutations at LRP binding site was amplified using primer set HS34FP/HS34RP. The forward primer HS34FP was designed by introducing mutations in the consensus LRP binding site (TATTTTTT). The conserved (TATTTTTT) bases found in the LRP binding site were changed to GGAAGAGA. While introducing these changes, the −10 and −35 hexamers were left unaltered. The amplicon obtained was sequenced to confirm the presence of mutations in LRP binding site before cloning it into promoter probe vector. The resulting abcR-lacZ with mutated LRP binding site was named as pHS13.
Insertion of ORF coding mCherry at the intergenic region of lutD and lutE
To clone ORF coding mCherry at the intergenic region of lutD and lutE, entire lut operon including promoter elements was amplified using PS1FP/PS1RP primer set and cloned in T-vector, pMD20. The resulting recombinant plasmid, pPS1 was then used as template to engineer XhoI and NheI sites at the intergenic region of lutD and lutE. Site-directed mutagenesis was performed by overlap extension PCR (79) using Phusion polymerase (Thermo Fisher Scientific India Pvt Ltd). Initial round of mutagenesis was performed by using pPS1 as template and PS2FP/PS2RP as primers. After ascertaining the creation of XhoI site, the second round of site-directed mutagenesis was performed using primer set PS3FP/PS3RP to create NheI site downstream of XhoI site. The resulting plasmid designated as pPS2 was used to insert ORF coding mCherry as XhoI and NheI fragment, the resulting plasmid pPS3 contains entire lut operon along with ORF coding for mCherry between lutD and lutE. The mCherry coding lut operon was then cloned in shuttle vector pPS4 as BamHI and SalI fragment. The generated plasmid pPS5 was then electroporated into abcR200 positive (DS002) and negative (HS002) strains of Acinetobacter baumanii.
Confocal microscopy
A. baumannii DS002 (pPS5) and HS002 (pPS5) cells were grown in minimal medium having 5 mM leucine and lactate as sole source of carbon and harvested before mid-log phase. The cells were washed in PBS buffer and resuspended in the same buffer to obtain final A600 of 5. A 10 μl drop was sealed with a cover slip on a glass slide. The cells were then observed under the super resolution STED microscope (Leica) (laser power-2%, HYD gain-100, PMT-450, objective-100X, and zoom factor-1.90).
Expression and purification of AbLRPC6His
E coli BL21 (pHS10) cells were grown to mid-log phase and the expression of _Ab_LRP^C6His^ was induced following standard procedures (67). After induction, the cells were lysed, and the clear lysate was obtained after centrifugation at 13,000 rpm. The lysate was then used to detect presence of _Ab_LRP^C6His^ by performing Western blot using anti-His antibodies. The _Ab_LRP^C6His^ found in the lysate was purified using metal ion affinity chromatography standardized in our laboratory (80) and the purity of _Ab_LRP^C6His^ was ascertained by analyzing the purified protein on SDS-PAGE (12. 5%).
Generation of lrp null mutant of E. coli AM001
lrp gene from E.coli AM001 was deleted following established procedures (81). Phage particles obtained after infecting lrp keio mutant of E.coli (82) were used to delete lrp gene from E.coli AM001. Colonies obtained after transduction were tested for deletion of lrp gene by performing colony PCR using the oligos HS35FP and HS35RP as primers. The lrp deletion derivative of E. coli AM001 strain was named as E. coli HS001.
LRP-abcR200 interactions: Two plasmid assay
A two-plasmid assay was developed to assess the role of Lrp on expression of abcR200. The E. coli HS001 strain was cotransformed with pHS12 (abcR200–lacZ fusion) and pHS11 coding _Ab_Lrp^C6His^ from an inducible promoter. The E. coli HS001 (pHS11+ pHS12) cells were then used to determine _Ab_Lrp^C6His^ dependent repression on the expression of abcR200. The E. coli HS001 (pHS11+ pHS12) cells were grown in the presence of IPTG to induce the expression of _Ab_Lrp^C6His^, and the promoter activity of abcR200 was assayed by measuring the β-galactosidase activity following the standard procedures (66). The E. coli HS001 containing either pHS12 or pHS11 along with E. coli HS001 (pHS11+ pHS12) cells grown in the absence of IPTG served as controls. While assessing the influence of leucine on abcR200 promoter activity, the E. coli HS001 (pHS11+ pHS12) cells were grown in minimal medium containing 0. 5, 1.0, 1.5, and 2.0 mM of exogenous leucine and without leucine. When the culture reached to mid-log phase, the cells were harvested and β-galactosidase activity was measured following standard procedures (66). E. coli HS001 (pHS11+ pHS12) cells grown in the absence of exogenous leucine served as control culture. Results of three independent experiments were used to perform statistical analysis using GraphPad Prism software. Data are presented as mean ± SD, and the p value of < 0.05 was considered statistically significant.
Leucine induced oligomerization of AbLRPC6His
The affinity purified _Ab_LRP^C6His^ was dialyzed against 20 mM Hepes, 150 mM NaCl, pH 8.0 buffer for 12 h. After dialysis 23 μM protein was taken in a clean eppendorf tube and the process of cross-linking was initiated by adding 1.2 μl (3 mM) DSS (Thermo Fisher Scientific) dissolved in dimethyl sulfoxide. The reaction mix was incubated at room temperature for 30 min to ensure good cross-linking among LRP proteins. When required either leucine (5 mM) or DNA (300 ng) containing LRP binding site was added to the reaction mix. The cross-linking reaction was stopped by the addition of quenching solution (1M Tris–Cl, pH-7.5) to a final concentration of 20 mM. Pure protein without any cross-linker served as control. These samples were analyzed on SDS-PAGE (12.5%), and Western blots were performed using anti-His antibodies to detect the oligomeric state of _Ab_Lrp^C6His^. The His-tagged protein mix loaded along with the samples served as molecular weight markers.
Generation of abcR200 KOs
The abcR200 was deleted in A. baumannii DS002 by replacing it with kanamycin cassette. As illustrated, the abcR200 gene overlaps with the omt gene and is transcribed in the opposite direction (Fig. 1A). The promoter of omt gene is in the middle of abcR200 gene and hence its deletion invariably disrupts omt gene promoter region. Briefly, upstream and downstream regions (500 bps) flanking abcR200 gene were amplified as NotI-BamHI and BamHI–SalI fragments using primers HS4FP/HS4RP and HS5FP/HS5RP, respectively. The purified PCR products were then digested with BamHI and ligated to generate a single NotI-SalI fragment. The kanamycin resistance cassette was then inserted at BamHI site before ligating it as NotI and SalI fragment into a suicidal vector (pJQ210) digested with similar enzymes (83). The resulting recombinant plasmid, pHS2 was used to transform E. coli S17-1. Biparental mating using E. coli S17-1 (pHS2) as a donor and A. baumannii DS002 as recipient was performed following standard procedures, and selection of exconjugants was done on LB plates containing kanamycin (84). The exconjugants were then screened for sucrose sensitivity (5% sucrose plates), and the colonies that lost vector backbone were selected to perform colony PCR using primer set HS4FP and HS5RP. The colony that contained kanamycin cassette in place of abcR200 gene was designated as A. baumannii HS002.
Transcriptomics
Transcriptome analysis of A. baumannii DS002 and abcR200 deletion derivative, A. baumannii HS002 was performed to identify the abcR200 responsive mRNAs. Three biological replicates of A. baumannii DS002 and A. baumannii HS002 were grown in carbon rich (LB) medium under identical conditions till late log phase. The cells were harvested, and the total RNA was isolated following the procedures mentioned elsewhere (85). Prior to library preparation, ∼2.5 μg of total RNA was depleted of rRNA using Ribo-Zero rRNA Removal Kit (Bacteria). About ∼50 ng of Qubit quantified ribo-depleted RNA was taken for fragmentation and priming. The fragmented and primed mRNA was further subjected to first strand synthesis in the presence of actinomycin D (Gibco, Life Technologies) followed by second strand synthesis. The double stranded cDNA was purified using HighPrep PCR magnetic beads (MAGBIO Genomics Inc). The purified cDNA was end-repaired, adenylated, and ligated to Illumina multiplex barcode adapters as per NEBNext Ultra Directional RNA Library Prep Kit protocol. The adapter ligated cDNA was purified using HighPrep beads and subjected to 14 cycles of indexing PCR (37 °C) for 15 min followed by denaturation at 98 °C for 30 s, and cycling (98 °C for 10 s, 65 °C for 75 s, and 65 °C for 5 min) to enrich the adapter-ligated fragments. The final PCR product (sequencing library) was purified with HighPrep beads and was subjected to library quality control check. The Illumina-compatible sequencing library was initially quantified by Qubit fluorometer (Thermo Fisher Scientific) and its fragment size distribution was analyzed on Agilent TapeStation. The libraries generated were subjected to Illumina sequencing. Raw sequencing reads were processed to remove the adapter sequences and the low-quality bases using Trimmomatic (v0.38) (http://www.usadellab.org/cms/?page=trimmomatic) (86) software with the following settings in paired end mode. LEADING/TRAILING:10 (1% error), SLIDINGWINDOW:5:16 (window size: quality ∼ 2.5% error), MINLEN:30. FastQC program was used to check read quality before and after the data cleaning (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Complete read count table is provided in the Table S1. Data cleaning and analysis of differential expression was performed on a cloud genomics platform (Stanome Pvt. Ltd).
Differential gene expression analysis
Reference genome and annotation (Genbank ID: CP027704.1) was downloaded from Genbank. Associated gff3 file with all the gene features was also obtained from the same source. Read abundance quantification was performed using salmon tool (v 0.11.2) (87). Normalized and filtered read counts were used for identifying the DEGs using the DESeq2 (88) tool with default settings. DEGs with a q-value of ≤ 0.10 and a log2 fold-change of ≥ 1 (upregulated) or log2 fold-change of ≤ 1 (downregulated) were selected as significant DEGs for further analysis. A logarithmic plot was generated to visualize the differentially expressed genes based on the expression pattern and set cutoff value for significance (Fig. S1).
RT PCR
Total RNA was isolated from bacterial cells using TRIzol Reagent (Sigma-Aldrich) following the manufacturer’s protocol. The concentration of the RNA isolated was determined spectrophotometrically using a Nanodrop. Total RNA isolated was converted into cDNA using Verso cDNA synthesis kit (Thermo Fisher Scientific) following the manufacturer’s protocol. The reactions were stored in −20 °C until further use. RT- PCR was performed using lutP specific forward (HS12 FP) and lutE (HS11 RP), lutD (HS7 RP), and lutR (HS10 RP) specific reverse primers to verify if the lut genes are organized as an operon in the genome of A. baumannii DS002. The RT-PCR was performed following established protocols (89).
Quantitative PCR
Quantitative PCR (qPCR) was performed to quantify the expression of lut-specific mRNAs following standard procedures. The 16S rRNA gene served as an internal control for calibrating the expression of other genes. The qPCR reactions were carried out in Applied Biosystems 7500 Real-Time PCR System operating with ABI 7500 software (https://www.thermofisher.com/mx/es/home/technical-resources/software-downloads/applied-biosystems-7500-real-time-pcr-system.html) using 20 ng of cDNA template, 0.25 μM primers, and 1X Takara SYBR Green master mix. The real-time expression analysis of each gene was carried out in triplicates, and the relative expression of genes were calculated using 2-ΔCt method (90). The expression levels of lut-specific mRNAs were quantified following standard procedures. Since deletion of abcR200, disrupted omt gene, we have complemented HS002 strain with ectopically expressed O-methyltransferase before isolating total RNA. The A. baumannii DS002 and HS002 (pHS14) cultures were grown to mid-log phase in a medium containing lactate as sole source of carbon. The total RNA isolated from these cultures was used to quantify the expression levels of lutD, lutR, lutE, and lutP genes by performing qPCR.
Quantification of lutP expression in A. baumannii HS002 (pHS16)
Initially, the abcR200 gene along with its indigenous promoter was amplified and ligated in pYH206 to generate recombinant plasmid pHS16 using infusion PCR cloning kit. The plasmid pHS16 was then electroporated into A. baumannii HS002 cells to generate abcR200 positive genetic background. The A. baumannii HS002 (pHS16) cells were then grown to mid-log phase in minimal media and used for isolating total RNA. The lutP specific transcript was then quantified by performing qPCR by following procedures described elsewhere (90).
Construction of expression plasmids
The lutR, lutE, and lutP genes were amplified as NdeI and XhoI fragments using primer pair showed in Table 2 and independently ligated in pET23b. The recombinant plasmids designated as pHS3, pHS4, and pHS5, code LutR^C6His^, LutE^C6His^, LutP^C6His^, respectively. These recombinant plasmids were then transformed into BL21, and their expression was induced following standard procedures. After confirming their expression in E. coli, the plasmid pHS3 was used as a template to amplify lutR as SmaI and SalI fragment and ligated in pHY206 digested with similar enzymes, and the resulting recombinant plasmid was designated as pHS6. Similarly, plasmids pHS4 and pHS5 served as a template to amplify lutE and lutP genes as EcoRI and SalI fragments. These fragments were then ligated independently in pHY206 to generate broad host range expression plasmids pHS7 and pHS8. These (pHS6, pHS7, and pHS8) plasmids were then independently mobilized into A. baumannii DS002 and HS002 strains and ectopically expressed LutR^C6His^, LutE^C6His^, and LutP^C6His^ were monitored by performing Western blots using anti-His antibodies. Similar strategy was followed while expressing omt gene in A. baumannii HS002 strain. The omt gene cloned in pHY206 as EcoRI fragment generated broad host range expression plasmid pHS14 and it codes for Omt^C6His^. After checking its expression in E. coli, the plasmid pHS14 was mobilized into A. baumannii HS002. Expression of Omt in HS002 (pHS14) was induced by adding IPTG (1 mM) to the cultures grown to mid-log phase and its expression was monitored by performing Western blots using anti-His antibodies.
ChIP assay
ChIP analysis was performed using lrp negative E.coli HS001 cells. Initially, the E. coli HS001 cells were transformed with two compatible plasmids. One of them (pHS11) codes _Ab_Lrp^C6His^ from an IPTG inducible promoter and the second plasmid (pHS12) carries abcR200-lacZ fusion. Control E. coli HS001 cells were generated either by replacing _Ab_Lrp^C6His^ coding expression plasmid with expression vector, pMMB206 or by introducing abcR200-lacZ fusion (pHS13) with a mutant Lrp binding site. The cultures were grown to mid-log phase either in leucine-free minimal medium or in a minimal medium supplemented with 2 mM and 5 mM leucine before inducing the expression of _Ab_Lrp^C6His^ by adding 1 mM IPTG. After inducing the expression of AbLrp^C6His^, the ChIP assays were performed by following the procedure described elsewhere (91). Briefly, cells were incubated with 1% formaldehyde for 10 min at 37 °C to facilitate crosslinking between interacting AbLrp^C6His^ and PabcR200. After crosslinking, the cells were lysed by adding the lysis buffer (1% SDS+ 10 mM EDTA+50 mM Tris, pH 8.1) and briefly sonicated to facilitate fragmentation of DNA. The cross-linked lysates were then diluted using dilution buffer (0.01% SDS+ 1.2 mM EDTA+ 16.7 mM Tris, pH 8.1 + 167 mM NaCl) and immunoprecipitated overnight at 4 °C using either anti-His or anti-IgG antibodies. Next, Protein A/G beads were added to the immunoprecipitated complex, then placed on shaker and incubated at 4 °C with mild shaking. After incubation, the beads were thoroughly washed with different wash buffers (low salt buffer, high salt buffer, and LiCl buffer) to remove nonspecifically bound antigens to protein A/G beads. The antibody-bound DNA-protein complexes were then eluted using elution buffer (1% SDS + 0.1 M sodium bicarbonate) and incubated at 65 °C for 4 h after adding 5 M NaCl to remove cross-links formed between protein and DNA. After de-cross-linking is completed, the samples were treated with proteinase K, and the degraded proteins were removed by performing phenol–chloroform extraction. Finally, the DNA present in the aqueous phase was precipitated and used as template to amplify PabcR200 using specific primers listed in Table 2.
Data availability
All the data presented in this document can be found within the manuscript and accompanying supplementary files.
Supporting information
This article contains supporting information.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Fournier P.E.Richet H.The epidemiology and control of Acinetobacter baumannii in health care facilities Clin. Infect Dis.4220066926991644711710.1086/500202 · doi ↗ · pubmed ↗
- 2Nocera F.P.Attili A.R.De Martino L.Acinetobacter baumannii: its clinical significance in human and veterinary medicine Pathogens 1020211273351370110.3390/pathogens 10020127 PMC 7911418 · doi ↗ · pubmed ↗
- 3Wong D.Nielsen T.B.Bonomo R.A.Pantapalangkoor P.Luna B.Spellberg B.Clinical and pathophysiological overview of acinetobacter infections: a century of challenges Clin. Microbiol. Rev.3020174094472797441210.1128/CMR.00058-16PMC 5217799 · doi ↗ · pubmed ↗
- 4Antunes L.C.Visca P.Towner K.J.Acinetobacter baumannii: evolution of a global pathogen Pathog. Dis.7120142923012437622510.1111/2049-632X.12125 · doi ↗ · pubmed ↗
- 5Bergogne-Bérézin E.Towner K.J.Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features Clin. Microbiol. Rev.91996148165896403310.1128/cmr.9.2.148PMC 172888 · doi ↗ · pubmed ↗
- 6Manchanda V.Sanchaita S.Singh N.Multidrug resistant acinetobacter J. Glob. Infect Dis.220102913042092729210.4103/0974-777X.68538 PMC 2946687 · doi ↗ · pubmed ↗
- 7Howard A.O'Donoghue M.Feeney A.Sleator R.D.Acinetobacter baumannii: an emerging opportunistic pathogen Virulence 320122432502254690610.4161/viru.19700 PMC 3442836 · doi ↗ · pubmed ↗
- 8Göttig S.Gruber T.M.Higgins P.G.Wachsmuth M.Seifert H.Kempf V.A.Detection of pan drug-resistant Acinetobacter baumannii in Germany J. Antimicrob. Chemother.692014257825792483375110.1093/jac/dku 170 · doi ↗ · pubmed ↗