The ART of Resistance
Ananya Ray, Namita Surolia

TL;DR
This study explores how mutations in the malaria parasite Plasmodium falciparum lead to resistance to the drug artemisinin, focusing on the role of autophagy in parasite survival.
Contribution
The study experimentally confirms that autophagy is a key survival mechanism in artemisinin-resistant parasites.
Findings
ART-induced ER stress upregulates parasite autophagy via the unfolded protein response pathway.
Resistant parasites show elevated autophagy protein expression compared to wild-type parasites.
The autophagy inhibitor MRT68921 has a decreased IC50 in resistant parasites, confirming autophagy's role in resistance.
Abstract
Recent emergence and spread of artemisinin (ART) resistance in South-east Asia caused by mutations in P. falciparum Kelch13 in the background of other mutations including mutations in the macroautophagy/autophagy-related protein PfATG18, has intensified studies towards understanding the molecular mechanisms of resistance. The autophagy pathway of the parasite has been hypothesized to engage in resistance-associated proteostasis involving enhanced phosphatidylinositol-3-phosphate vesiculation, oxidative stress, unfolded protein response and also reduced hemoglobin endocytosis resulting from nutrient-limiting conditions, albeit without any experimental evidence. We demonstrate that ART-induced ER stress leads to upregulation of parasite autophagy through the unfolded protein response pathway. In addition, we show elevated basal expression of autophagy proteins in the ART resistant…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
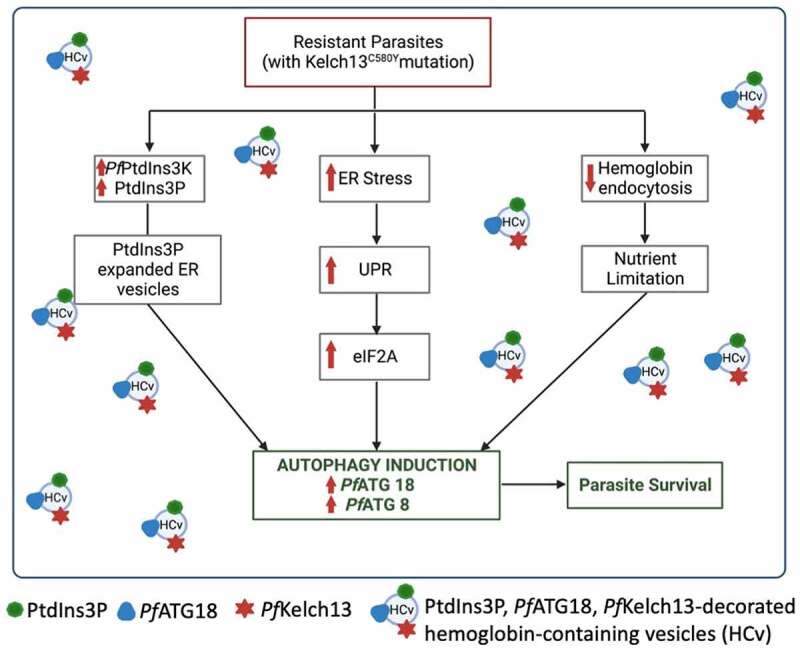 Figure 1
Figure 1 Figure 2
Figure 2- —Department of Biotechnology (DBT), Distinguished Biotechnology Research Professorship
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutophagy in Disease and Therapy · Mosquito-borne diseases and control · Malaria Research and Control
Artemisinin-based combination therapies have significantly reduced the malaria burden and deaths globally. These first-line treatments combine a long-acting partner drug (pyronari-dine, sulfadoxine-pyrimethamine, mefloquine, lumefantrine, piperaquine) with a fast-acting and effective antimalarial derivative of artemisinin (ART) such as artesunate, artemether and dihydroartemisinin (DHA). Malaria parasites have, however, begun to develop resistance to ART, posing a threat to the eradication of the disease. The diminishing effectiveness of artemisinin-based combination therapies against P. falciparum raises concerns that ART resistance may evolve and spread to malaria-endemic regions. Current understanding of the mechanisms of P. falciparum by which parasites eventually develop resistance to ART is limited.
ART induces widespread cellular damage by alkylating proteins promiscuously, resulting in the buildup of misfolded proteins in the parasite ER and cytoplasm. This results in proteopathy leading to disruption of various parasite metabolic functions and viability. Protein homeostasis, involving proper protein folding and vesicular remodeling (proteostasis), as well as reduced hemoglobin endocytosis are the two major mechanisms proposed to be responsible for ART resistance. Several point mutations in PfKelch13 in association with mutations in other proteins such as the autophagy-related protein PfATG18 have been identified as the genetic markers for ART resistance, with the PfKelch13^C58^°^Y^ mutation being the most prevalent. It has been suggested that mutations in PfKelch13 mitigate ART-induced proteopathy through proteostatic dysregulation of phosphatidylinositol 3-kinase, leading to elevation of phosphatidylinositol-3-phosphate (PtdIns3P) and subsequent expansion of homeostatic ER-PtdIns3P vesicles of proteostasis. Amplified PtdIns3P vesicles disseminate proteostatic capacity throughout the parasite, which may prevent ART-induced parasite death. Reduced hemoglobin endocytosis during the early ring stage of the parasite’s asexual cycle also forms the basis for the PfKelch13-mediated ART resistance. Studies have provided evidence that PfKelch13 is also involved in hemoglobin endocytosis in the parasite. The major mutation in PfKelch13 impairs PfKelch13 levels, which significantly reduces hemoglobin endocytosis and degradation. The decreased availability of hemoglobin confers ART resistance as hemoglobin-derived iron is necessary to activate ART. Though each of these pathways, as well as PtdIns3P vesiculation, can independently induce autophagy, a direct demonstration for the involvement of autophagy in resistance is lacking.
Autophagy is a self-degradative process, engaged in various stress responses such as starvation, hypoxia and oxidative stress; providing macromolecules to recycle and mediating cell homeostasis. Autophagy proteins function in concert to maintain protein quality control by degrading misfolded proteins in the lysosome of eukaryotic cells. ER stress-mediated activation of unfolded protein response (UPR) signaling transducers regulates autophagy at various stages, including induction, phagophore nucleation, and phagophore expansion. Along with a robust ER stress response mechanism, the parasite genome also encodes a small number of partially conserved autophagy-related proteins that have been implicated in autophagy-like processes, it is, however, not known whether the parasite autophagy pathway would be activated in response to ER stress. Additionally, a specific mutation (T38I) in PfATG18 is strongly selected under ART resistance and confers a fitness advantage to parasites by allowing for faster growth rates under nutrient-limited conditions. Our findings show that 2517 P. falciparum genome isolates from Southeast Asia and Africa share the T38I mutation in PfATG18 and the C580Y mutation in PfKelch13 [1]. The wild-type PfATG18 is also thought to be phosphorylated at the T38 position. PfATG18 binds to and utilizes PtdIns3P for membrane association. We speculate that the mutation in PfATG18 could be selected during ART resistance to compensate for the parasite fitness loss associated with resistance by increasing its association with PtdIns3P, thereby promoting autophagy in the parasite. Involvement of parasite autophagy in proteostasis mechanisms of ART resistance to a large extent remains unreported.
In our recent work, we investigated the cross-talk between ER stress, UPR and the autophagy-like pathway in the malaria parasite. We found that DHA (the active ART metabolite)- mediated proteostatic stress increases eIF2A phosphorylation, reducing protein translation and promoting dormancy. Live cell imaging with PfSEC62-GFP, an ER membrane-localized parasite protein, reveal that the parasite ER does expand following DHA exposure, an indication of ER stress. Induction of autophagy upon DHA treatment is demonstrated by an increase in the number of PfATG8-labeled autophagosome-like vesicles and the expression levels of two parasite autophagy-related proteins, PfATG8 and PfATG18. Inhibition of PK4-eIF2A-mediated UPR signaling decreases PfATG8 and PfATG18 expression levels, further validating our proposition that ER stress-induced autophagy is mediated through the UPR.
Further we provide novel insights into the role of parasite autophagy in ART resistance mechanisms. The transcript and the protein levels of PfATG8 and PfATG18 are higher in the ART-resistant Kelch13^C58^°^Y^ isolate as compared to its isogenic WT Kelch13 isolate. Also, the resistant parasites show increased colocalization of PfATG18-labeled vesicles with the autophagosome marker PfATG8 as compared to the isogenic one, indicating activated autophagy in the parasites even at the basal level. Amino acid starvation further activates autophagy in both WT Kelch13 and Kelch13^C58^°^Y^ isolates, although the colocalization of PfATG18 and PtdIns3P-labeled vesicles is increased in ART-resistant parasites compared to isogenic parasites. A dose-response analysis of the specific autophagy inhibitor MRT68921 indicates a 2-fold reduction in the inhibitor’s IC50 in ART-resistant parasites compared to its isogenic counterpart. Importantly, we find that PfKelch13, along with PfATG18, is transported to the food vacuole via hemoglobin-containing vesicles, which transport host-derived hemoglobin to the parasite food vacuole.
Altogether, we find a previously unexplored interplay between ART-mediated ER stress, UPR, and autophagy in P. falciparum, as well as the contribution of parasite autophagy to the establishment of ART resistance (Figure 1). Our findings thus highlight the potentials of inhibiting the parasite autophagy as a target to develop antimalarials for resistant P. falciparum infection. Figure 1.Autophagy mediates ART resistance in P. falciparum.
The reference list from the paper itself. Each links out to its DOI / PubMed record.