Molecular Structure of the mRNA Export Factor Gle1 from Debaryomyces hansenii
Min Jeong Jang, Soo Jin Lee, Jeong Ho Chang

TL;DR
This study reveals the crystal structure of a truncated Gle1 protein from Debaryomyces hansenii, offering insights into its role in mRNA export.
Contribution
The paper presents the first high-resolution structure of unbound Gle1 from yeast, providing structural insights into its function.
Findings
The DhGle1ΔN protein has 13 α-helices and was crystallized at 1.5 Å resolution.
Structural comparisons showed no major conformational changes but identified distinct secondary structural elements in specific helices.
The study contributes to understanding Gle1's architecture and its interactions with Dbp5 during mRNA export.
Abstract
Gle1 functions as a regulator of Dbp5, a DEAD-box-containing RNA helicase that is a component of the nuclear pore complex. In association with Gle1 and inositol hexakisphosphate (IP6), ADP-bound Dbp5 facilitates the release of RNA. The RNA-bound Dbp5 undergoes ATP hydrolysis and is activated by Gle1 in the presence of IP6. The formation of a ternary complex involving Dbp5, Gle1, and the nucleoporin Nup159 promotes ADP secretion and prevents RNA recombination. To date, several complex structures of Gle1 with its binding partners have been described; however, the structure of unbound Gle1 remains elusive. To investigate the structural features associated with complex formation, the crystal structure of N-terminally truncated Gle1 from Debaryomyces hansenii (DhGle1ΔN) was determined at a resolution of 1.5 Å. The DhGle1ΔN protein comprises 13 α-helices. Structural comparisons with homologs,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
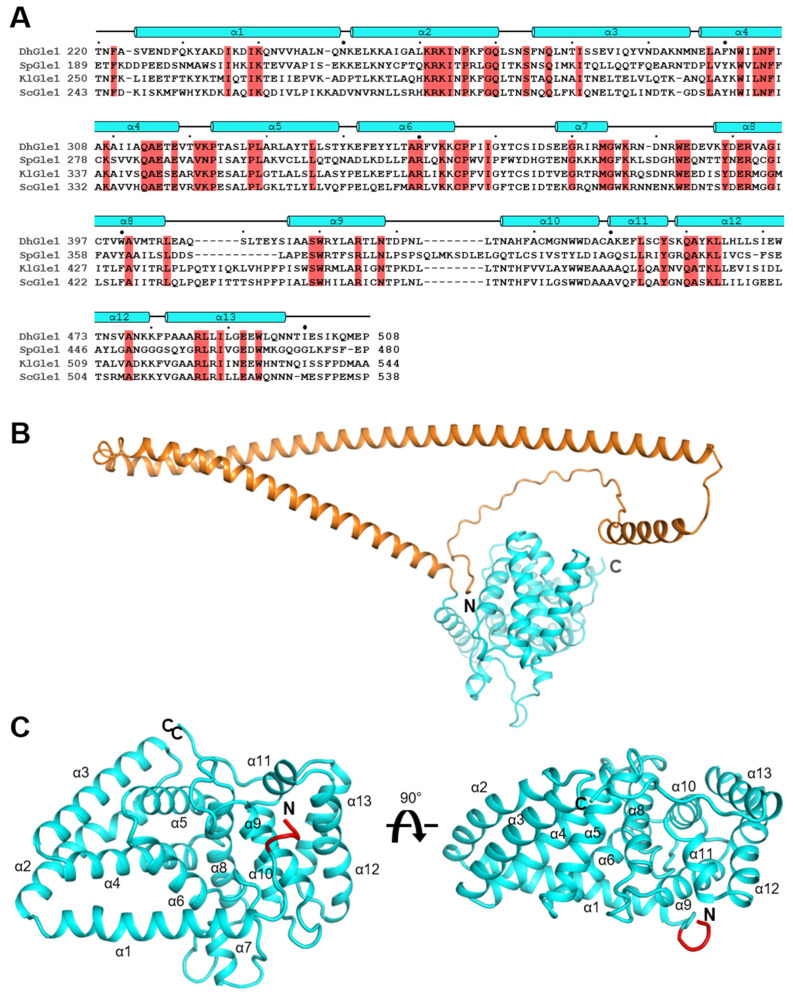 Figure 1
Figure 1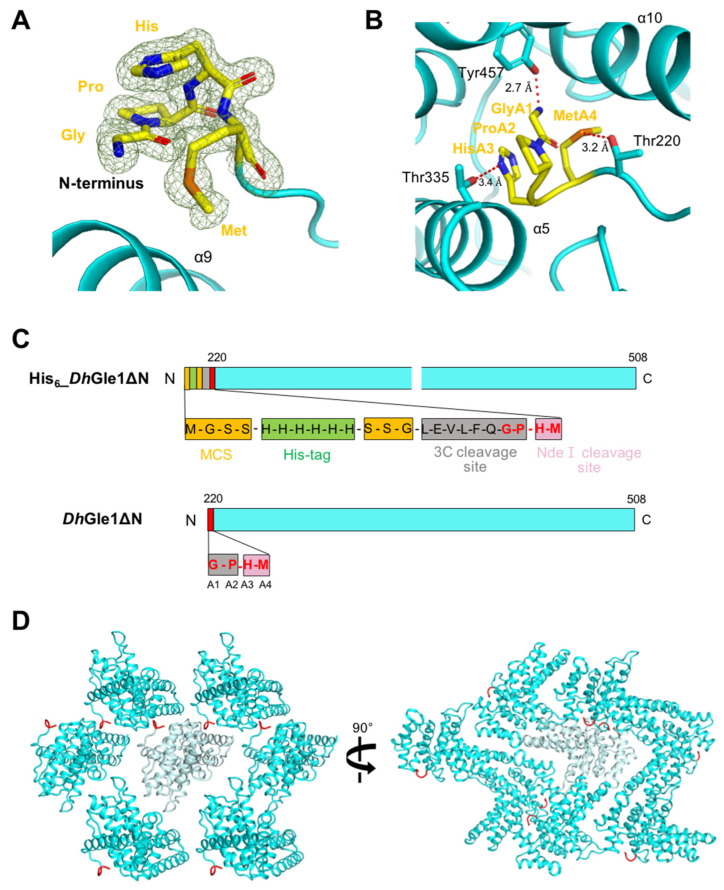 Figure 2
Figure 2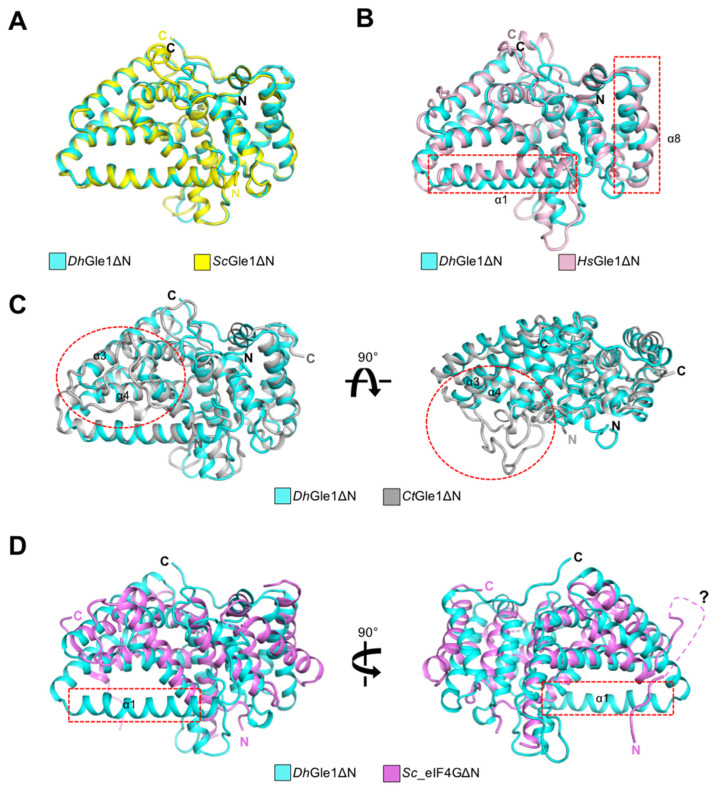 Figure 3
Figure 3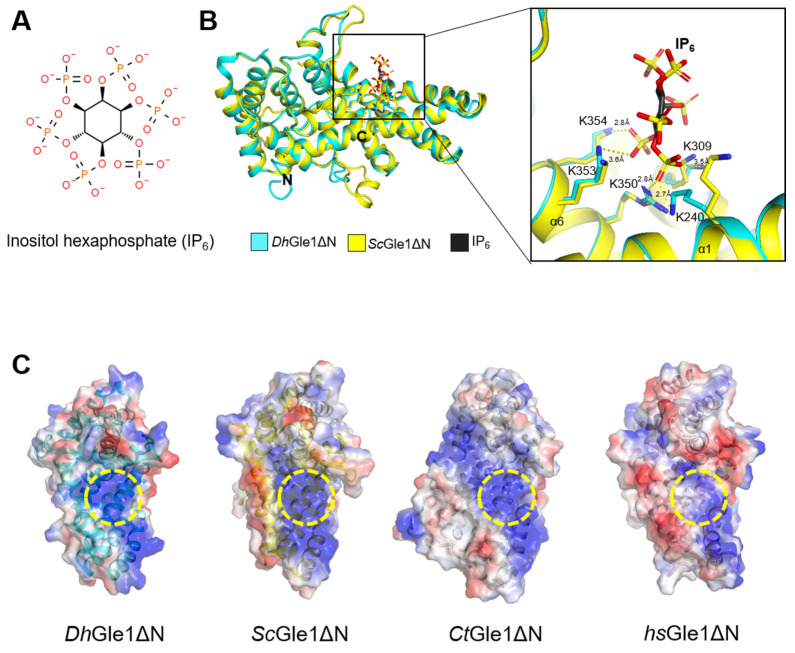 Figure 4
Figure 4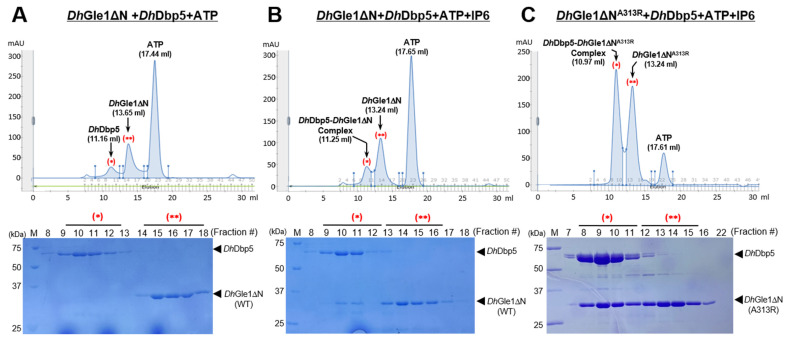 Figure 5
Figure 5- —Ministry of Science and ICT
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA Research and Splicing · Nuclear Structure and Function · RNA and protein synthesis mechanisms
1. Introduction
In eukaryotic cells, transcribed mRNA must be exported from the nucleus through nuclear pores for subsequent translation in the cytoplasm [1,2,3]. This export process requires the precise selection of mRNA by the nuclear pore complex (NPC) to ensure the exclusion of aberrant mRNAs [4,5]. Within the nucleus, properly processed mRNA interacts with various proteins in the NPC during export. During this process, certain mRNA-bound proteins are prevented from being transported by the RNA helicase DEAD-box protein 5 (Dbp5), which is located in the cytoplasmic region of the NPC [4,6]. A pivotal aspect of mRNA export is the dissociation of proteins from the messenger ribonucleoprotein complexes (mRNPs), which is believed to play a role in directing the export [2,4]. Consequently, only mRNAs are released into the cytoplasm. However, the regulation of mRNA remodeling remains not completely understood [2,4,6].
The superfamily 2 (SF2) RNA helicase Dbp5 is a DEAD-box protein found in a wide range of prokaryotic and eukaryotic organisms [2,4,6,7]. Dbp5 binds to RNAs in an ATP-dependent manner [8]. ATP facilitates the interaction between two distinct domains of Dbp5: the N-terminal domain (NTD) and the C-terminal domain (CTD), which in turn regulates RNA binding activity. Moreover, within the NPC filaments, Dbp5 is essential for the dissociation of the mRNA export receptor Mex67 and the RNA-binding protein Nab2 from the mRNP complex, a process that is dependent on its ATPase activity [2,8,9]. The Dbp5–Gle1–Nup159 complex orchestrates mRNA export in the NPC, utilizing various factors, including ATP, ADP, and the mRNP [2,4,6,10]. The RNA-bound Dbp5 acts through ATP hydrolysis, a process facilitated by both Gle1 and the small molecule inositol hexakisphosphate (IP6) [2,4,11,12]. A recent report indicated that the ATPase activity of Dbp5 is not activated by either tRNA or double-stranded RNA alone; rather, it necessitates the concurrent presence of Gle1 for activation [13].
Gle1 interacts with both the NTD and the CTD of Dbp5. The presence of ADP-bound Dbp5, in conjunction with Gle1 and IP6, facilitates RNA release by promoting the separation of these two domains. When this separation occurs, the Dbp5-NTD engages with the N-terminal region of Nup159, further enhancing the disconnection of the two domains [14]. The formation of the ternary complex consisting of Dbp5, Gle1, and Nup159 not only promotes the secretion of ADP but also prevents RNA recombination and encourages enzyme recycling [6,15]. While Nup159 is not associated, it facilitates the dissociation of ADP from Dbp5 within the NPC by inducing a conformational change in Dbp5 [15]. Although the structure of Gle1 and its ternary complex with Dbp5 and Nup159 has been elucidated in Saccharomyces cerevisiae, the precise mechanism underlying the mRNA export cycle remains not completely understood [4,6].
To investigate the Dbp5-mediated mRNA export cycle, we focus on the structure and function of Gle1. Several structures of Gle1 have been reported to date, including its binary complex with Dbp5 and its ternary complexes with both Dbp5 and Nup142, as well as Dbp5 and Nup159 [3,6,16]. However, the structure of Gle1 alone has yet to be characterized. To better understand the structural features associated with complex formation, we determined the crystal structure of Gle1 from Debaryomyces hansenii at a resolution of 1.5 Å. This structure was extensively compared to those of its structural homologs, including eIF4G. The findings of this study may provide insights into the molecular structure of Gle1 in fungal species, thereby expanding our understanding of its function.
2. Results
2.1. Overall Structure of DhGle1ΔN
Expression of full-length Gle1 in Escherichia coli leads to inclusion body formation. To obtain a protein suitable for crystallization, we constructed a truncated form of Debaryomyces hansenii Gle1 (residues 220–508, DhGle1ΔN) in which the N-terminal region was removed from the protein (Figure 1A). The excluded N-terminal region is comprising three α-helices containing two long helices based on a predicted full-length Gle1 structure that possibly interacts with several nucleoporins other than Dbp5, and does not fully interact with Dbp5 [3,17,18,19] (Figure 1B).
The structure of DhGle1ΔN belongs to the space group P2_1_2_1_2_1_, and there is one molecule in the asymmetric unit. The monomeric DhGle1ΔN structure consists of 13 α-helices (Figure 1C), revealing that Gle1 comprises an all-α-helical HEAT repeat protein that interacts with both RecA-like domains of Dbp5 [6].
Interestingly, an extra N-terminal segment consisting of four residues—Gly-Pro-His-Met—was clearly shown in the electron density (Figure 2A). These residues, labeled A1–A4, were stabilized by Thr220, Thr335, and Tyr457 through hydrogen bonds (Figure 2B). Specifically, GlyA1, HisA3, and MetA4 interact with Tyr457, Thr335, and Thr220, respectively. To investigate the origin of this extra N-terminal segment, we analyzed the modified pET28a vector containing a 3C protease cleavage site. This analysis revealed that the multiple cloning site (MCS) of the vector includes a hexahistidine tag, the 3C cleavage site (Leu-Glu-Val-Leu-Phe-Gln-/Gly-Pro), and an Nde I restriction site (His-Met) (Figure 2C, upper panel). Upon cleavage of the histidine tag by the 3C protease, the four residues Gly-Pro-His-Met remained (Figure 2C, lower panel). Thus, while His6_DhGle1ΔN contained a 23-amino acid artificial N-terminal segment, DhGle1ΔN featured only the four additional N-terminal residues present in the untagged recombinant protein.
To explore the presence of the N-terminal extra segment in the structure, we analyzed the crystallography packing status. Each segment of the monomer is stabilized by neighboring molecules, fitting well into the space between them (Figure 2D, Supplementary Figure S1). In contrast, the His6_DhGle1ΔN molecules did not pack tightly together in the crystal structure due to their shape and the presence of the longer N-terminal segment including the hexahistidine tag, resulting in a poor X-ray. The crystallographic packing pattern suggests that the extended N-terminal segment may inhibit stable interactions between adjacent molecules. Taken together, these findings indicate that the N-terminal tag plays a critical role in determining the quality of the crystals.
2.2. Structural Comparison of DhGle1ΔN with Its Homologs
Homologous structures of DhGle1ΔN were identified using the DaLi web-based server, which facilitated systematic structural comparisons with molecules in the Protein Data Bank [20]. The top three identified homologs were Gle1 in complex with Dbp5 from S. cerevisiae (ScGle1ΔN; PDB ID: 3PEV), Gle1 in complex with Nup42 GBM from Homo sapiens (HsGle1ΔN; PDB ID: 6B4F), and Gle1 in complex with Nup42 GBM from Chaetomium thermophilum (CtGle1ΔN; PDB ID: 6B4H), with root mean square deviations (r.m.s.d.) of 1.2, 2.3, and 2.2 Å, and Z scores of 41.1, 29.8, and 29.5, respectively (Table 1). The three homologous Gle1 structures were compared based on their equivalent regions in relation to DhGle1ΔN. Notably, DhGle1ΔN displayed structural similarity to the eukaryotic translation initiation factor eIF4G, which serves as a platform for the SF2 DEAD-box ATPase eIF4A from S. cerevisiae (Sc_eIF4GΔN in complex with Sc_eIF4A, PDB ID: 2VSX), showing a root mean square deviation (r.m.s.d.) of 3.9 Å and a Z score of 15 (Table 1).
Overall, DhGle1ΔN and the four homologous structures exhibit relatively similar folding patterns Among these, the fold of ScGle1ΔN is nearly identical to that of DhGle1ΔN, with the exception of local loops, including the N- and C-termini, resulting in an r.m.s.d. of 1.2 Å (Figure 3A, Table 1). Although DhGle1ΔN and HsGle1ΔN share similar structural characteristics, distinct conformations were observed at the α1-helix and α8-helix (Figure 3B). When comparing DhGle1ΔN to CtGle1ΔN, the loop connecting the α3-helix and α4-helix in CtGle1ΔN, which comprises 30 residues, is significantly longer than that in DhGle1ΔN (Figure 3C). On the other hand, the structure of Sc_eIF4GΔN diverges more markedly from that of DhGle1ΔN. Firstly, the α1-helix present in DhGle1ΔN is absent in Sc_eIF4GΔN; this α1-helix plays a crucial role in stabilizing the cofactor IP6. Secondly, the region spanning from the α8-helix to the α13-helix demonstrates particularly poor superimposition due to the structural differences between the two proteins. Lastly, the conformation of the N-terminal region in Sc_eIF4GΔN is entirely distinct from that of DhGle1ΔN (Figure 3D). A portion of the N-terminal loop (14 residues) in Sc_eIF4GΔN is not visible due to its inherent flexibility. Consequently, while Sc_eIF4GΔN does not interact with IP6, this flexible N-terminal region may potentially compensate to the IP6-independent interaction with eIF4A [6] (Figure 3D). Given its structural similarity to Sc_eIF4GΔN, it is plausible that DhGle1ΔN also plays a role in interactions with RNA helicase activators.
2.3. IP6 Binding Sites in DhGle1ΔN and Homologous Proteins
The negatively charged small molecule IP6 mediates the binding of Gle1 to Dbp5 [21] (Figure 4A). To explore the residues involved in the interaction between IP6 and DhGle1ΔN, the structure of DhGle1ΔN was superimposed with the Gle1–Dbp5 complex from S. cerevisiae (PDB ID: 3PEU) (Figure 4B). The basic residues Lys240, Lys309, Arg350, Lys353, and Lys354 in DhGle1ΔN interact with IP6 via hydrogen bonds or salt bridges. As expected, the IP6 binding pocket in DhGle1ΔN is highly basic, similar to those in ScGle1ΔN and CtGle1ΔN, whereas HsGle1ΔN displays a less positively charged pocket (Figure 4C). This basic pocket is conserved across fungal species but is not present in humans. Moreover, it has been demonstrated that IP6 is not required in the association of HsGle1ΔN with Dbp5 [3].
2.4. IP6-Dependent Complex Formation of DhGle1ΔN with DhDbp5
To evaluate the interaction between DhGle1ΔN and DhDbp5, size-exclusion chromatography (SEC) analyses were conducted. The SEC profiles showed that the main peaks for DhGle1ΔN and DhDbp5 appeared at elution volumes of 13.67 mL and 11.26 mL, respectively. To investigate complex formation, DhGle1ΔN and DhDbp5 proteins were incubated with ATP for 30 min at 4 °C, followed by SEC analysis. Based on the SEC peak profiles and SDS-PAGE results, the two proteins did not interact under these conditions (Figure 5A). The SDS-PAGE analysis clearly showed that DhGle1ΔN and DhDbp5 remained distinct, with separation observed in lanes 9 to 18. The elution volumes for DhDbp5, DhGle1ΔN, and ATP were 11.16 mL, 13.65 mL, and 17.44 mL, respectively. Subsequently, we mixed DhGle1ΔN and DhDbp5 in the presence of both ATP and IP6 for further SEC analysis (Figure 5B). The SDS-PAGE results from SEC fractions 8 to 18 indicated that the bands of DhDbp5 and DhGle1ΔN co-migrated in fractions 9 to 11 (Figure 5B, lower panel). The peak elution volumes for the DhDbp5-DhGle1ΔN complex, DhGle1ΔN, and ATP were 11.25 mL, 13.24 mL, and 17.65 mL, respectively. However, a comparison of the SEC profiles for DhDbp5 and the DhDbp5–DhGle1ΔN complex revealed an inconsistency, as DhDbp5 eluted earlier than the DhDbp5–DhGle1ΔN complex (Figure 5A,B). Additionally, the ATP peaks also exhibited differences between the two SEC profiles. This led us to hypothesize that IP6 might influence the elution volumes by altering the buffer conditions or inducing the protein’s conformational change. Furthermore, the band intensity of DhGle1ΔN in the DhDbp5 complex fraction on the SDS-PAGE indicated a weak interaction between the two proteins. Consequently, we performed SEC analysis using a gain-of-function mutation of DhGle1ΔN, specifically altering the 313rd Alanine to Arginine (DhGle1ΔN^A313R^), which corresponds to the 337th Histidine to Arginine in Gle1 from S. cerevisiae based on sequence alignment (Supplementary Figure S2) [6]. As anticipated, the SDS-PAGE gel results confirmed that the bands for DhDbp5 and DhGle1ΔN^A313R^ migrated together clearly (Figure 5C). Additionally, the peak positions for DhGle1ΔN^A313R^ and ATP were comparable to those observed for DhGle1ΔN and ATP in the SEC profile (Figure 5B,C). Significantly, the peak position for the DhDbp5-DhGle1ΔN^A313R^ complex shifted forward during elution, indicating that DhDbp5 and DhGle1ΔN^A313R^ form a more stable complex than DhDbp5 and DhGle1ΔN in the presence of IP6.
3. Discussion
The protein Gle1, in conjunction with Dbp5 and Nup159, plays a crucial role in the ATP- and IP6-dependent export of mRNA [22]. In this study, we aimed to explore the structural properties of D. hansenii Gle1 by revealing its crystal structure and extensively comparing it with those of homologous proteins. Initially, we used the full-length DhGle1 for structural characterization; however, this approach proved unsuccessful due to the protein’s local flexibility. Based on a previous report from the Weis group, we engineered a truncated version of the protein, excluding the N-terminal 219 residues (His6_DhGle1ΔN), which allowed for successful crystallization [6]. Despite this improvement, the crystals diffracted poorly, making it difficult to determine the structure. Following a series of optimizations, we produced a further modified version of DhGle1 lacking the 19-residue N-terminal segment containing the His6-tag (DhGle1ΔN), which diffracted exceptionally well at a resolution of 1.5 Å. This finding highlighted that the additional N-terminal segment from the artificial cloning tag was critically involved in the crystallographic packing and significantly enhanced the quality of the diffraction data obtained from the crystals.
The structures of Sc_eIF4GΔN and DhGle1ΔN display notable similarities, yet their functions diverge significantly [6]. Specifically, Gle1 and eIF4G interact with the RNA helicases Dbp5 and eIF4A, respectively. These binding factors serve as platforms to stimulate RNA helicase activity [23]. This raises the question: why do these cells possess two similar cofactors in the cytoplasm? To address this, it is essential to examine the role and localization of the RNA helicases. Firstly, Dbp5 functions as a shuttle between the nucleus and cytoplasm, primarily residing on the cytoplasmic side of the NPC in association with Nup159 [4]. Because Dbp5 is located at the cytoplasmic face of the NPC, it can facilitate the release of mRNA from the nucleus into the cytoplasm upon Gle1 binding [24]. In contrast, eIF4A exists in the cytoplasm, forming a complex with eIF4G that is integral to translation initiation. Secondly, although the structures of Sc_eIF4G and DhGle1 are largely similar, there are several variations in their local conformations. Notably, the α1-helix of DhGle1 is absent in Sc_eIF4G, and there are distinct differences in the regions spanning from the α8 helix to the α13 helix between the two structures (Figure 3).
In the SEC analysis, neither DhDbp5 nor DhGle1ΔN exhibited binding in the presence of ATP and MgCl_2_. However, upon the addition of IP6, the two proteins formed a complex, although SEC analysis indicated that this interaction was weak (Figure 5). What accounts for the weak IP6-dependent interaction between DhGle1ΔN and DhDbp5? First, it is worth noting that the DhDbp5 used in the SEC analysis was the full-length protein. The N-terminal region of Dbp5 may influence its interaction with DhGle1ΔN. To support the speculation, the structure of the N-terminal 111 residues of DhDbp5 was predicted using AlphaFold-2 [19]. This analysis revealed that the N-terminal region of DhDbp5 is predominantly unstructured, comprising only two short helices (Supplemental Figure S3). Consequently, this flexible and unstructured N-terminal region might destabilize the interaction with DhGle1ΔN. Further supporting this assumption, the Weis group used a truncated Dbp5 that excluded the N-terminal 90 residues, along with gain-of-function mutations in Saccharomyces cerevisiae Dbp5 and Gle1 (ScDbp5^L327V^ and ScGle1^H337R^), to establish the Dbp5-Gle1-ADP-IP6 complex [6,8]. Furthermore, DhGle1^A313R^, corresponding to ScGle1^H337R^, was shown to form a stable complex in the presence of IP6. Notably, Dbp5 can adopt two distinct conformations, depending on whether it is bound to ADP or ATP [6,15]. Hence, it is likely that ADP-bound Dbp5 establishes more stable interactions with DhGle1ΔN. Taken together, further investigations, including experiments using N-terminally truncated DhDbp5, ADP-bound DhDbp5, and mutations in both DhDbp5 and DhGle1, will be necessary to establish a more stable complex between DhGle1ΔN and DhDbp5 for structural studies.
Despite our advanced knowledge of mRNA export mechanisms, the intricate molecular details of these processes across diverse species remain not fully understood. To fulfill this knowledge gap, it is crucial to obtain a stable ternary complex of Gle1, Dbp5, and Nup159 from fungal species. Only by achieving this we can determine the structures of the complex, which will enable us to clarify how DhGle1ΔN recognizes and interacts with DhDbp5 and DhNup159. In future studies, given that Dbp5 also plays a role in the nuclear export of pre-ribosomal subunits and tRNAs, it will be important to investigate Gle1’s potential involvement in these processes as well [25,26]. Furthermore, exploring other aspects of nuclear export would be advantageous, as it is currently unclear which proteins are removed from the mRNP during mRNA release and how mRNA length influences the duration of the export process.
4. Materials and Methods
4.1. Cloning and Overexpression of Gle1
The gene encoding Gle1 in D. hansenii (NCBI ID: 2903325) was obtained from genomic DNA, and an N-terminally truncated construct was referred to as DhGle1ΔN (220–508). These genes were amplified via PCR using the forward primer 5′-AAGGCATATGACTAATTTTGCTTCTGTTGAAAA-3′ and reverse primer 5′-AAG GCTCGAGCTATGGTTCCATTTGCTTAATT-3′. The amplified fragments were digested with restriction enzymes NdeI and XhoI (Ezynomics; Daejeon, Republic of Korea), and then the digested fragments were ligated into pET28a vectors containing a 3C protease cutting site using T4 ligase (M0202S; Roche, Mannheim, Germany). The recombinant plasmids were then transformed into E. coli strain DH5α and confirmed by DNA sequencing. To obtain the site-directed mutant A313R, a site-specific mutation was created by PCR-based methods using Phusion high-fidelity DNA polymerase (ThermoFisher; Waltham, MA, USA) with the forward primer 5′-AAG GCT ATA ATA CGT CAA GCA G-3′ and the reverse primer 5′-CTG CTT GAC GTA TTA TAG CCT T-3′. The recombinant plasmid was sequenced to confirm correct incorporation of the mutations.
4.2. Purification of Recombinant Proteins
The recombinant DhGle1ΔN-pET28a-3C and DhGle1ΔN^A313R^-pET28a-3C plasmids were transformed into BL21(DE3) star cells for overexpression. Cells were grown in Luria–Bertani medium (Ambrothia, Daejeon, Republic of Korea) containing 50 mg/L kanamycin (Applichem, St. Louis, MO, USA). After reaching an optical density of 0.6, cells were induced with 0.3 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 20 °C for 18 h. The harvest cells were resuspended in a buffer containing 20 mM Tris (pH 8.0; Sigma-Aldrich, St. Louis, MO, USA), 250 mM NaCl (Applichem, St. Louis, MO, USA), 5% glycerol (Affymetrix, Santa Clara, CA, USA), 0.2% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA), 10 mM β-mercaptoethanol (BioBasic, Markham, ON, Canada), and 0.2 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich, St. Louis, MO, USA). Cells were lysed by ultrasonication (VCX-500/750; Sonics & Materials, Inc., Newtown, CT, USA) with 3 s on/off cycles continuously for 20 min. Cell debris was removed by centrifugation at 13,000 rpm for 40 min, and then the supernatant was loaded into an Ni-NTA HiTrap chelating column (GE Healthcare, Mississauga, ON, Canada) with a flow rate of 3 mL/min. After washing with a buffer (50 mM Tris, pH 8.0, 200 mM NaCl) containing 20 mM imidazole (Sigma-Aldrich, St. Louis, MO, USA), the proteins were eluted with a buffer (50 mM Tris, pH 8.0, 200 mM NaCl) containing 500 mM imidazole (Sigma-Aldrich, St. Louis, MO, USA). To remove the His_6_-tag from the expressed proteins, 250 units of 3C protease were treated at 7 °C for 12 h. The cleaved His_6_-tag was removed by additional Ni-NTA affinity chromatography. The protein was further purified using a HiPrep 16/60 Sephacryl S-300 HR column (GE Healthcare, Mississauga, ON, Canada). A buffer used for SEC contained 20 mM Tris (pH 7.5), 150 mM NaCl, and 2 mM dithiothreitol (DTT; Calbiochem, Darmstadt, Germany). The purified proteins were more than 98% pure checked by SDS-PAGE. The final concentration of the protein was 26.7 mg/mL, and it was stored at −80 °C for subsequent experiments.
4.3. Crystallization
Preliminary crystallizations of DhGle1ΔN were performed under more than 400 conditions using MCSG crystallization screening solution kits (Molecular Dimensions Ltd., Calibre Scientific UK, Rotherham, UK) at 7 °C. Crystals grew under 3 conditions: (1) 0.1 M CHES (pH 9.5) and 30% (w/v) PEG 3000, (2) 0.1 M BICINE (pH 9.0) and 20% (w/v) PEG 6000, and (3) Tris (pH 8.5) and 25% (w/v) PEG 3350. These three conditions were used for additional screening. Within at most five days, thick polygon-shaped crystals were obtained in drops containing equal volumes (1 µL) of the protein sample (25 mg/mL) and reservoir solution. The crystals were preserved in a cryoprotectant solution containing crystallization buffer and 30% (v/v) glycerol. The samples were flash-frozen and stored in liquid nitrogen.
4.4. Data Collection and Structure Determination
Diffraction datasets were collected at 100 K on a beamline 5C using a Quantum 315 CCD detector (Area Detector Systems Corporation, Poway, CA, USA) at the Pohang Accelerator Laboratory (PAL; Pohang, Republic of Korea). Crystal structures were solved by the molecular replacement method using PHENIX software version 1.9 (https://phenix-online.org/; Lawrence Berkeley Laboratory, Berkeley, CA, USA) [27]. Structural models were built using Coot [28], followed by model refinement using the PHENIX refine program. The structures were visualized using PyMOL (https://pymol.org; Schrödinger Inc., New York, NY, USA). Statistics of the data collection and refinement processes are provided in Table 2. The PDB files of Gle1 and its complex have been uploaded to the DALI web server (http://ekhidna2.biocenter.helsinki.fi/dali/, accessed on 6 November 2022) for comparison with other homologous structures using the PDB search and pairwise tools.
4.5. Size-Exclusion Chromatography Anaysis
The purified DhGle1ΔN, DhGle1ΔN^A313R^, and DhDbp5 full-length proteins were subjected to size exclusion chromatography (SEC) with a buffer containing 25 mM Tris-Cl (pH 7.5), 150 mM NaCl, 2 mM DTT by using AKTA Pure (GE Healthcare, Mississauga, ON, Canada), respectively. To determine whether they form a complex, incubate DhGle1ΔN (30 μM) and DhDbp5 (30 μM) proteins in the presence of either 10 mM ATP or 10 mM ATP and 10 mM IP6 for 30 min at 4 °C. The proteins DhGle1ΔN^A313R^ (50 μM), and DhDbp5 (50 μM) were also incubated in the presence of 10 mM ATP and 10 mM IP6 for 30 min at 4 °C. The reacted proteins were further applied to the Superose-12 column (GE Healthcare, Mississauga, ON, Canada) for SEC analysis. A buffer used for SEC contained 25 mM Tris-Cl (pH 7.5), 150 mM NaCl, and 2 mM DTT.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tseng S.S. Weaver P.L. Liu Y. Hitomi M. Tartakoff A.M. Chang T.H. Dbp 5p, a cytosolic RNA helicase, is required for poly(A)+ RNA export EMBO J.1998172651266210.1093/emboj/17.9.26519564047 PMC 1170606 · doi ↗ · pubmed ↗
- 2Tran E.J. Zhou Y. Corbett A.H. Wente S.R. The DEAD-box protein Dbp 5 controls m RNA export by triggering specific RNA:protein remodeling events Mol. Cell 20072885085910.1016/j.molcel.2007.09.01918082609 · doi ↗ · pubmed ↗
- 3Lin D.H. Correia A.R. Cai S.W. Huber F.M. Jette C.A. Hoelz A. Structural and functional analysis of m RNA export regulation by the nuclear pore complex Nat. Commun.20189231910.1038/s 41467-018-04459-329899397 PMC 5998080 · doi ↗ · pubmed ↗
- 4Hodge C.A. Tran E.J. Noble K.N. Alcazar-Roman A.R. Ben-Yishay R. Scarcelli J.J. Folkmann A.W. Shav-Tal Y. Wente S.R. Cole C.N. The Dbp 5 cycle at the nuclear pore complex during m RNA export I: dbp 5 mutants with defects in RNA binding and ATP hydrolysis define key steps for Nup 159 and Gle 1Genes. Dev.2011251052106410.1101/gad.204161121576265 PMC 3093121 · doi ↗ · pubmed ↗
- 5Stewart M. Nuclear export of m RNA Trends Biochem. Sci.20103560961710.1016/j.tibs.2010.07.00120719516 · doi ↗ · pubmed ↗
- 6Montpetit B. Thomsen N.D. Helmke K.J. Seeliger M.A. Berger J.M. Weis K. A conserved mechanism of DEAD-box AT Pase activation by nucleoporins and Ins P 6 in m RNA export Nature 201147223824210.1038/nature 0986221441902 PMC 3078754 · doi ↗ · pubmed ↗
- 7Heung L.J. Del Poeta M. Unlocking the DEAD-box: A key to cryptococcal virulence?J. Clin. Investig.200511559359510.1172/JCI 2450815765144 PMC 1052016 · doi ↗ · pubmed ↗
- 8Weirich C.S. Erzberger J.P. Flick J.S. Berger J.M. Thorner J. Weis K. Activation of the D Ex D/H-box protein Dbp 5 by the nuclear-pore protein Gle 1 and its coactivator Ins P 6 is required for m RNA export Nat. Cell Biol.2006866867610.1038/ncb 142416783364 · doi ↗ · pubmed ↗