Short Scalable Route to Bis-morpholine Spiroacetals and Oxazepane Analogues: Useful 3D-Scaffolds for Compound Library Assembly
Daniel Kovari, Louise Male, Kimberley A. Roper, Christian P. Mang, Oliver Kunz, Liam R. Cox

TL;DR
This paper presents a scalable method to synthesize complex molecular scaffolds useful for drug discovery.
Contribution
A novel four-step synthesis of sp3-rich spiroacetal scaffolds with morpholine rings is introduced.
Findings
The method allows high-yield synthesis of spiroacetal scaffolds on a large scale.
The approach enables substitution of morpholine rings with oxazepanes to generate diverse analogues.
The resulting compounds occupy a drug-like chemical space but are structurally novel.
Abstract
sp3-Rich molecular scaffolds incorporating nitrogen heterocycles represent important starting points for assembling compound screening libraries and drug discovery. Herein, we report a four-step synthesis of a conformationally well-defined sp3-rich scaffold incorporating two morpholine rings embedded within a spiroacetal framework. The synthesis involves the intermediacy of a 2-chloromethyl-substituted morpholine, accessed from epichlorohydrin and readily available β-aminoalcohols. Base-mediated dehydrochlorination affords an exocyclic enol ether, from which the second morpholine ring is constructed in two steps. Scaffold synthesis is high-yielding and can be performed on a large scale. The methodology allows ready substitution of one–or both– of the morpholine rings for 1,4-oxazepanes and the generation of 6,7- and 7,7-spiroacetal analogues, which are virtually unexplored in drug…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
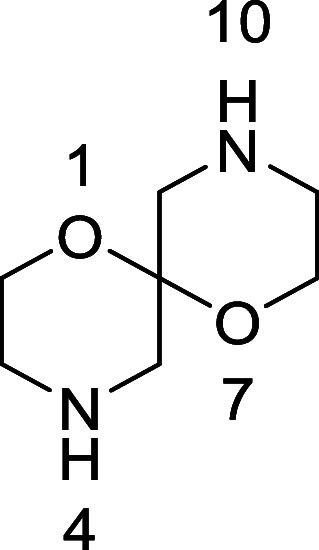 Figure 1
Figure 1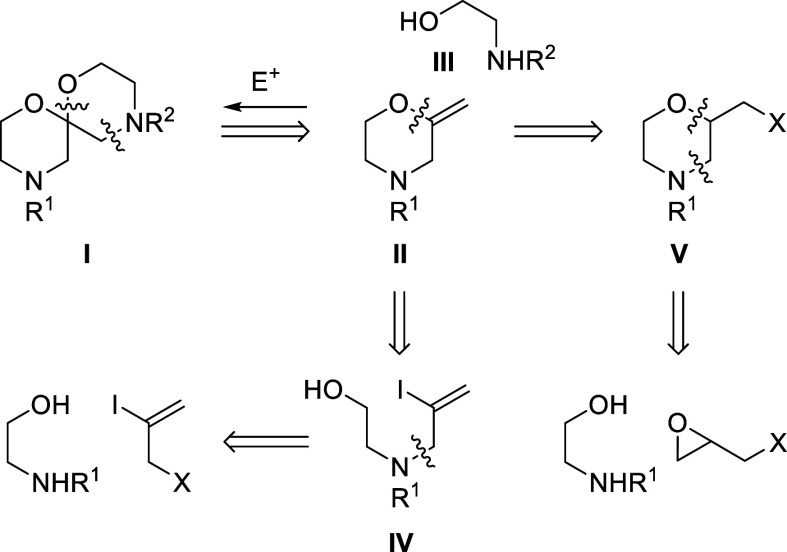 Scheme 1
Scheme 1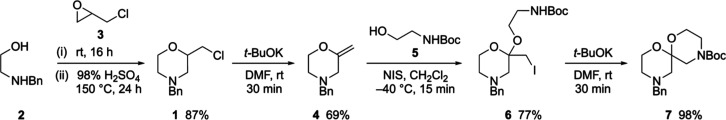 Scheme 2
Scheme 2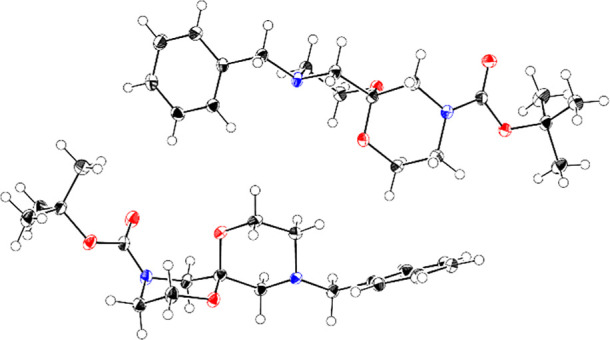 Figure 2
Figure 2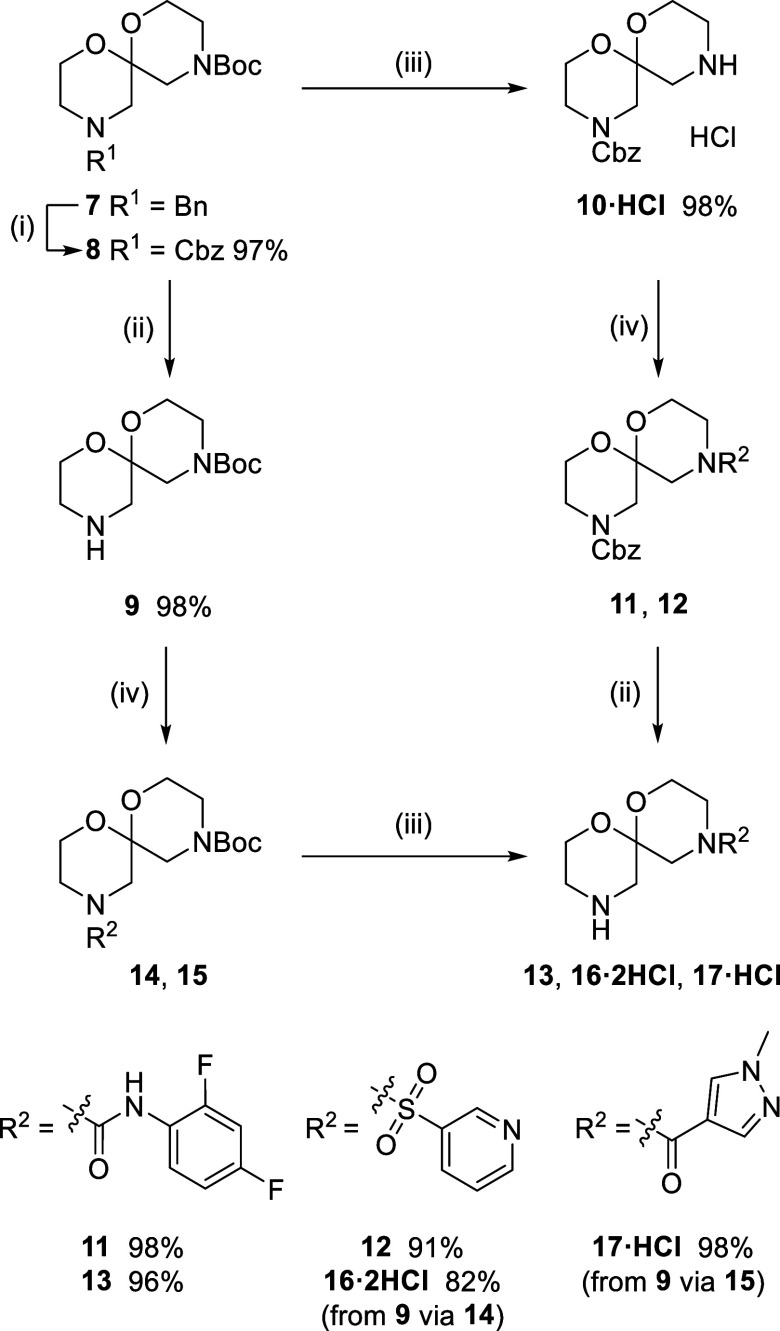 Scheme 3
Scheme 3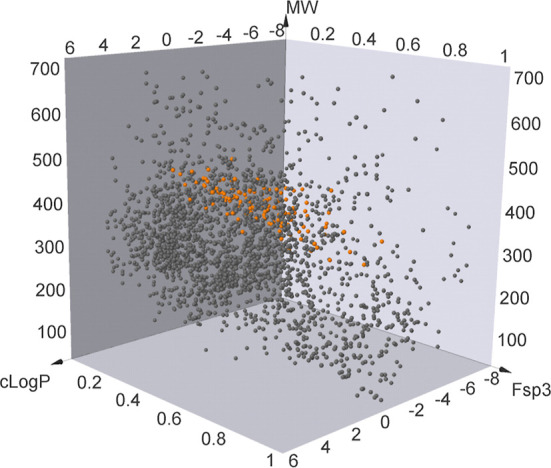 Figure 3
Figure 3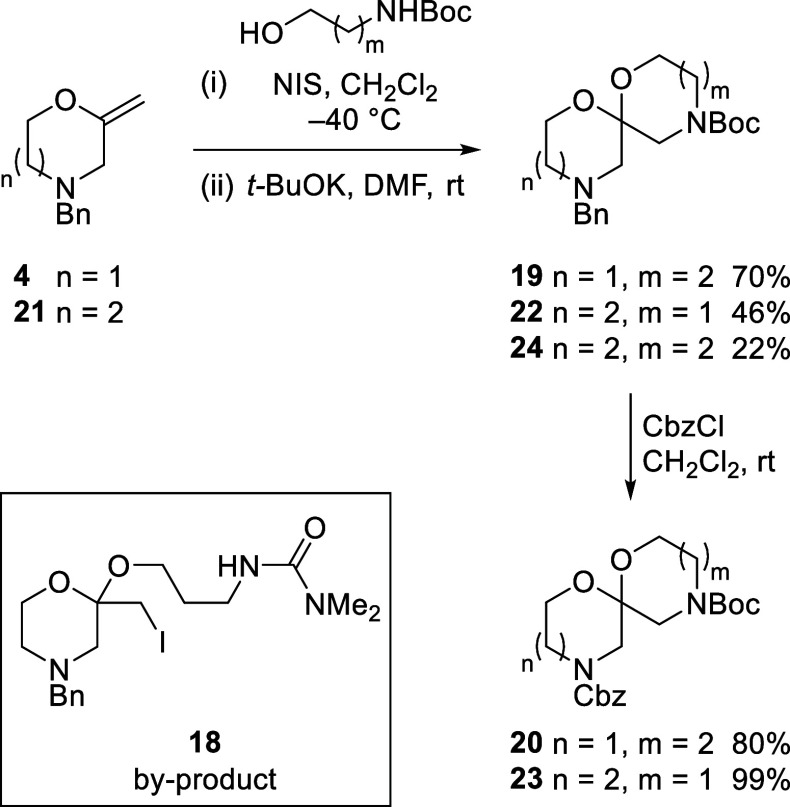 Scheme 4
Scheme 4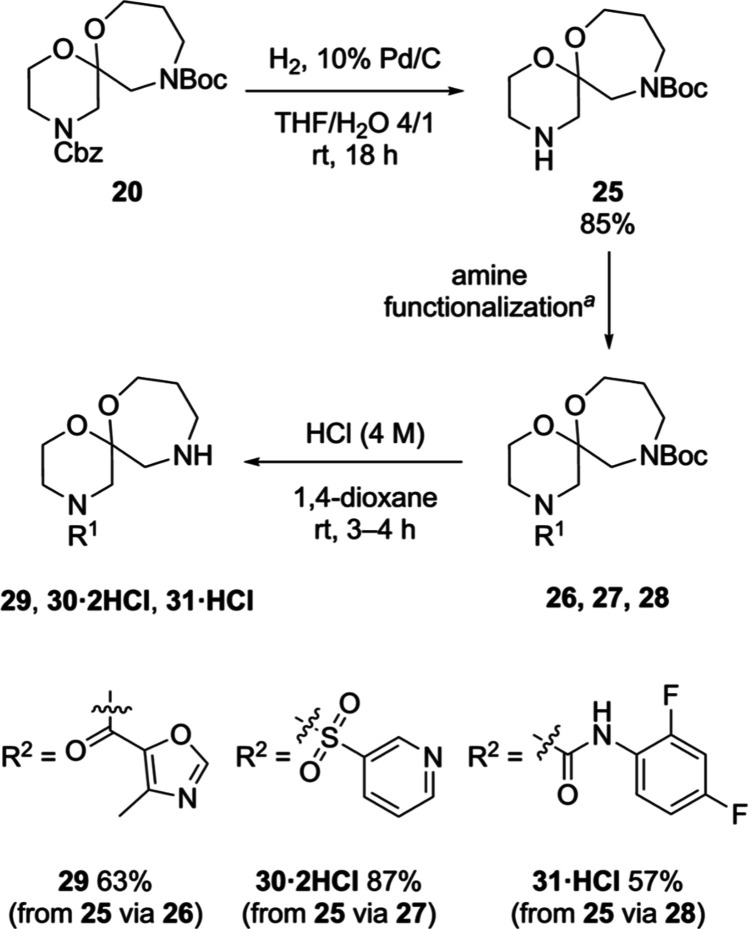 Scheme 5
Scheme 5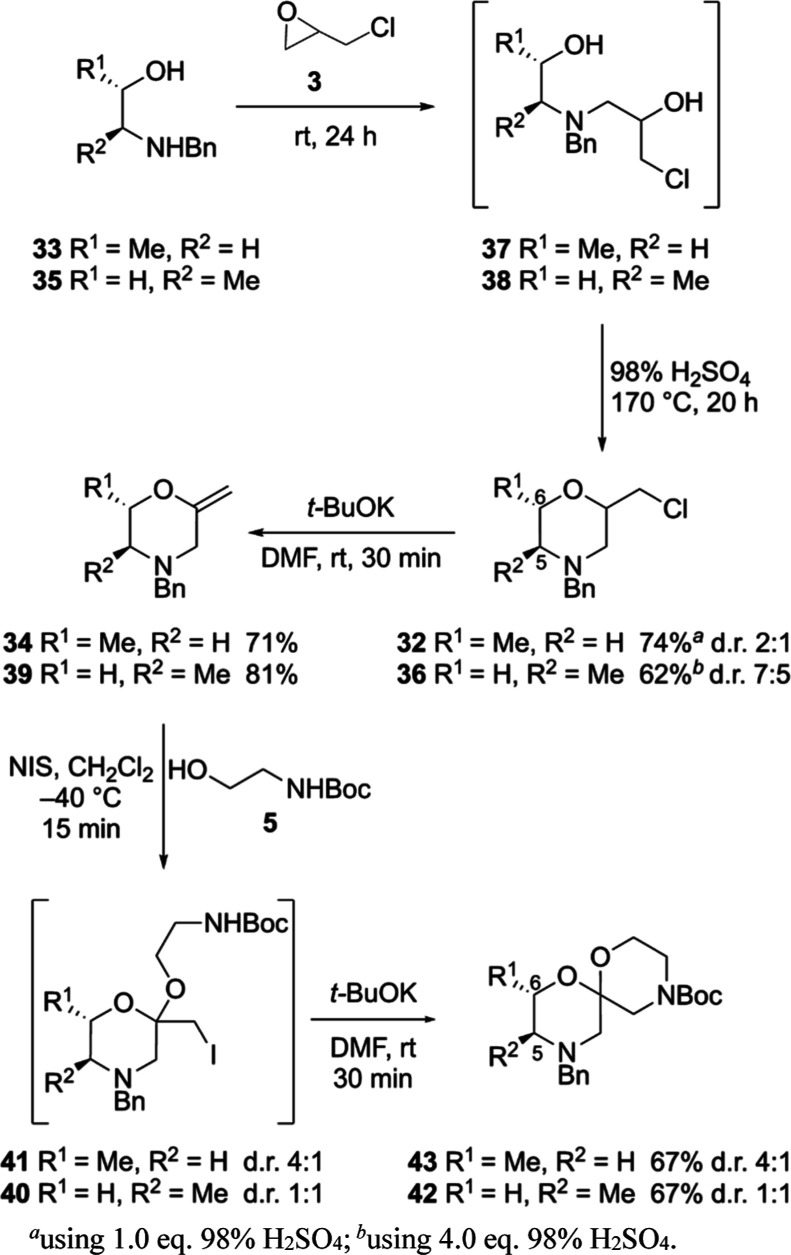 Scheme 6
Scheme 6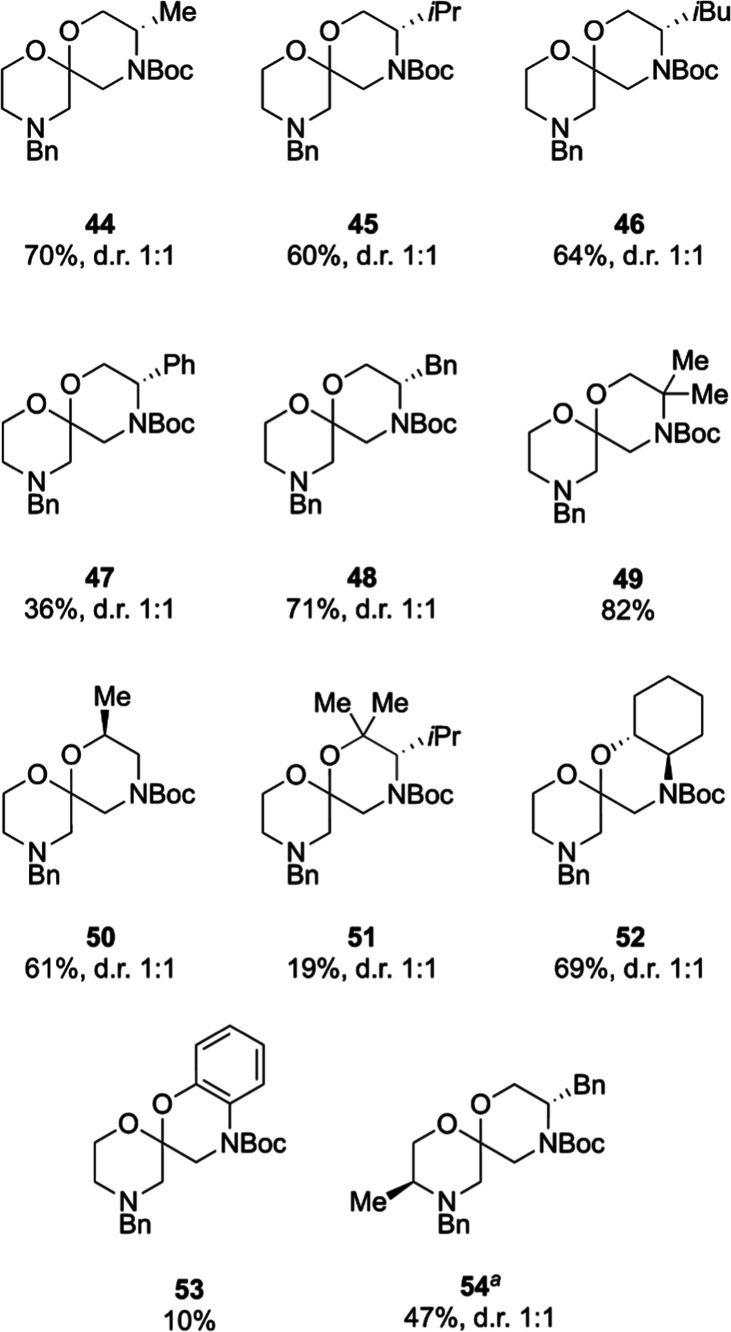 Figure 4
Figure 4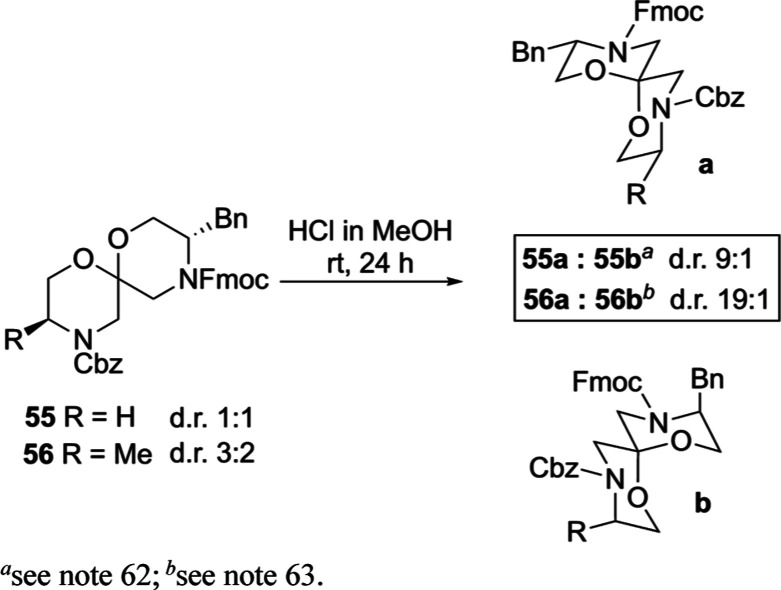 Scheme 7
Scheme 7- —H2020 Marie Sklodowska-Curie Actions10.13039/100010665
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalytic C–H Functionalization Methods · Synthetic Organic Chemistry Methods · Oxidative Organic Chemistry Reactions
Introduction
Conformationally well-defined three-dimensional scaffolds are attractive starting points for compound library assembly and applications in early-stage drug discovery. Conformational rigidity in these sp^3^-rich systems can be achieved in various ways, through bridged systems and ring fusion in polycycles,^1^ as well as through spiro systems. Spirocyclic molecules are widespread among bioactive natural products^2^ and have received growing interest as scaffolds for drug discovery.^3^ The presence of a spiranic center within the core of a molecular scaffold necessarily introduces a high degree of three-dimensionality, typically significant conformational rigidity, and a limited number of low-energy conformations. Functionality embedded within the constituent rings then introduces exit vectors and a means to predictably orient recognition motifs into well-defined regions of three-dimensional space, which is attractive for structure-based drug design.^3a,4^ Moreover, there are examples in which the incorporation of a spirocycle imparts favorable physicochemical properties^5^ and an improved pharmacokinetics profile compared with nonspirocyclic analogues.^3a^ A number of groups have reported diversity libraries of spiro(hetero)cyclic compounds^6^ and bis-spirocyclic frameworks.^7^ Müller and co-workers used a cheminformatics approach to classify systematically the structures and chemical space interrogated by spirocycles of known biological activity.^3b^ This study highlighted the capacity for this type of scaffold to access a significant volume of biologically relevant chemical space, reinforcing the attraction of deploying spirocycles for drug discovery.
Spiroacetals are an important class of spirocycle;^8^ they are ubiquitous across natural products, many of which display potent and diverse biological activities.^9^ While the hydrolytic instability of acyclic acetals might raise a safety flag owing to the aldehyde/ketone hydrolysis product, the increased stability of spiroacetals and their abundance in natural products justifies their inclusion in compound screening libraries and potential to deliver attractive starting points for drug discovery.^10^ Aliphatic nitrogen heterocycles also abound in bioactive natural products and are privileged structural motifs for drug discovery. The tetrahydro-1,4-oxazine–or morpholine– framework is particularly widespread, and synthetic routes to this nitrogen heterocycle, its prevalence in drugs, and pharmacological activity have been extensively reviewed.^11^ Azaspiro systems have been accessed in various ways,^12^ and azaspiro scaffolds incorporating a morpholine exhibit diverse bioactivities. Examples include AZD-2115, a dual-acting M3 receptor antagonist/β2-adrenergic receptor agonist,^3a^ NK1-antagonists,^13^ and potent HIV protease inhibitors,^14^ while a pyrrolomorpholine spiroacetal forms the core of the shensongine and acortatarin classes of natural products, which display antioxidant activity.^15^
Results and Discussion
We postulated that a spiro-bis-morpholine [1,7-dioxa-4,10-diazaspiro[5.5]undecane] framework (Figure 1) would provide an attractive scaffold for compound library assembly,^16^ especially if a scalable route could be developed that enabled sequential functionalization of the two amines. We envisaged orthogonally protected spiro bis-morpholine I could be accessed in two steps from 2-methylidenemorpholine II, using an electrophile-mediated regioselective functionalization of the exocyclic alkene with an ethanolamine bis-nucleophile III(17) to assemble the spiroacetal and second morpholine ring (Scheme 1). Two approaches to enol ether II were explored.
1,7-Dioxa-4,10-diazaspiro[5.5]undecane as a scaffold for compound library synthesis.
Retrosynthetic Analysis of Target Bis-morpholine Spiroacetal I
Stoltz and co-workers reported the synthesis of a range of 2-methylidenemorpholines, including enol ether II (R^1^ = Bn), via an intramolecular Ni(COD)2-catalyzed cross-coupling reaction of the corresponding iodo-aminoalcohol IV (Scheme 1).^18^ This reaction was successfully applied to a range of vinyl iodides (see the Supporting Information), including one containing a secondary amine [IV (R^1^ = H)], which extends the scope of this methodology; however, we were unable to access unsubstituted 2-methylidenemorpholine II (R^1^ = Bn) efficiently, achieving 20% conversion at best.^19^ A second approach was therefore explored, which ultimately provided a higher yielding, more practical, and scalable synthesis of this enol ether.
We envisaged enol ether II (R^1^ = Bn) could also be accessed via dehydrohalogenation^20^ of the corresponding 2-halomethyl-substituted morpholine V (Scheme 1).^21^ 2-Chloromethylmorpholine 1 was therefore prepared in a two-step, one-pot operation,^22^ first introduced by Loftus^22a^ and later improved by Matsumoto and co-workers;^22b^ thus, treatment of N-benzylethanolamine (2) with an equimolar quantity of epichlorohydrin (3) in the absence of solvent at room temperature, followed by the addition of 98% H_2_SO_4_ and heating at 170 °C, afforded 2-chloromethylmorpholine 1 in 87% yield (Scheme 2). While this reaction sequence employs particularly harsh conditions in the second step, it proved remarkably scalable and was performed on a 0.37 mol scale, providing 72 g of the target product. Subsequent dehydrochlorination on 1 using t-BuOK as the base,^23^ and DMF as solvent,^24^ afforded 2-methylidenemorpholine 4 (Scheme 2). Unlike the corresponding 2-methylidenetetrahydropyran, which needs to be stored over KOH,^25^ 2-methylidenemorpholine 4 could be purified by column chromatography using a basic eluent (see the Supporting Information for full details) and the purified product was stored under nitrogen at 4 °C for several weeks without any noticeable decomposition.^26^
Synthesis of Bis-morpholine Spiroacetal 7
Adapting a methodology introduced by Nishi and co-workers to assemble 2,2-disubstituted morpholines,^27^ 2-methylidenemorpholine 4 underwent regioselective iodoacetalization upon treatment with N-iodosuccinimide in the presence of N-Boc-ethanolamine (5) to give iodide 6. Subsequent treatment with t-BuOK in DMF effected ring closure to provide bis-morpholine spiroacetal 7. Starting from 17 g (92 mmol) of enol ether 4 delivered target scaffold 7 in 75% yield over the two steps (Scheme 2).^28^ The structural identity of 7 was confirmed by single-crystal X-ray diffraction, which revealed both rings adopt chair conformations and the spiroacetal benefits from double anomeric stabilization (Figure 2). A solution of 7 in DMSO left in an NMR tube at room temperature on an open bench over 3 weeks showed no evidence of decomposition, as determined by ^1^H- and ^13^C{^1^H}-NMR spectroscopy; this scaffold was therefore deemed sufficiently stable for compound library synthesis.
ORTEP plot of 7 with ellipsoids drawn at the 50% probability level, confirming the double anomeric stabilization of the spiroacetal. The structure contains two crystallographically independent molecules as shown. Atomic displacement parameters at 100 K.
Orthogonal deprotection of the two amines in spiroacetal 7 was next explored. Standard benzylamine deprotection conditions (H_2_/Pd/C in MeOH with or without acetic acid and the use of 1-chloroethyl chloroformate in the presence of acid scavengers) proved ineffective.^29^ As Cbz carbamates undergo hydrogenolysis more readily,^30^ benzylamine 7 was converted to carbamate 8 (Scheme 3).^31^ Now, hydrogenolysis of 8, performed on a 33 mmol scale, proceeded without event under neutral conditions^32^ to deliver secondary amine 9 with the Boc group intact.
Orthogonal Deprotection and Sequential Functionalization of Spiro-bis-morpholine 8Reaction conditions: (i) CbzCl, CH2Cl2, rt, 24 h; (ii) H2, 10% Pd/C, THF/H2O 4/1, rt; (iii) HCl (4 M in 1,4-dioxane), THF/H2O 4/1, rt; (iv) 2,4-difluoro-1-isocyanatobenzene, Et3N, CH2Cl2, rt, 1 h, or pyridine-3-sulfonyl chloride, Et3N, CH2Cl2, 0 °C–rt, 16 h, or 1-methyl-1H-pyrazole-4-carboxylic acid, HATU, iPr2NEt, CH2Cl2, 0 °C–rt, 16 h.
While a protecting group exchange is undesirable on efficiency grounds, the reaction sequence was straightforward and provided 9 in 95% yield over the two steps. The selective deprotection of the Boc carbamate in 8 was also confirmed: treatment with 4 M HCl in 1,4-dioxane^33^ led to rapid Boc deprotection, leaving the 6,6-spiroacetal intact, to provide 10, which was conveniently isolated as its HCl salt (Scheme 3).
Having access to two differently monoprotected bis-morpholine spiroacetals offered useful flexibility for scaffold decoration. Thus, treatment of amine HCl salt 10·HCl with 2,4-difluoro-1-isocyanatobenzene and pyridine-3-sulfonyl chloride under standard conditions provided urea 11 and sulfonamide 12, respectively (Scheme 3). Hydrogenolysis of the Cbz carbamate in urea 11 provided 13 in excellent yield; however, the same reaction with sulfonamide 12 saw recovery of the starting material, even when the carbamate deprotection was performed under acidic conditions to protonate the basic nitrogen in the embedded pyridine (Scheme 3). Turning instead to Boc carbamate 9, reaction with pyridine-3-sulfonyl chloride and 1-methyl-1H-pyrazole-4-carboxylic acid provided sulfonamide 14 and amide 15, respectively. This time, subsequent Boc deprotections proceeded without event, affording sulfonamide 16·2HCl in 82% yield and amide 17·HCl in 98% yield, over the two steps.
Having successfully functionalized one of the morpholine rings and deprotected the resulting products, the KNIME analytics platform^34^ was used to enumerate a virtual library of 630 compounds, from which 124 diverse^35^ compounds were selected for physical synthesis (see the Supporting Information). Compounds were chosen to occupy drug-like chemical space, obeying Lipinski’s rule of five and Veber’s rules.^36^ Urea 13 and 12 singly decorated scaffolds, synthesized from Boc carbamate 9 in yields ranging between 66 and 98% over the two steps, were applied in amidation, sulfonylation, urea formation, and reductive amination reactions to assemble a compound library (see the Supporting Information).^37^ Of the 124 reactions performed, only one failed to deliver any product after purification by HPLC, confirming bis-morpholine spiroacetal I as a viable scaffold for compound library assembly.^38^
To assess its chemical space coverage, the bis-morpholine spiroacetal library was compared with the collection of small-molecule drugs (molecular weight (MW) < 700 Da^39^) that have been approved by the Food and Drug Administration,^40^ in the fraction of sp^3^ carbon atoms (Fsp^3^)—lipophilicity (c Log P)—MW chemical space (Figure 3). The 3D scatter plot indicates that the library occupies similar physicochemical space to the FDA-approved list of small-molecule drugs. To evaluate the dissimilarity of the compound library from this set of FDA-approved drugs,^41^ Tanimoto coefficients^42^ were calculated (see the Supporting Information); the obtained scores of 0.2–0.4 indicate significant dissimilarity; thus, while our library compounds have similar physicochemical properties to clinically approved small-molecule drugs, they are structurally distinct. We hypothesize that they may act on new targets and, therefore, represent attractive starting points for drug discovery.
Comparison of library of synthesized spiro-bis-morpholine compounds (n = 124) with the FDA-approved small-molecule drugs39 (n = 2709) visualized in c Log P/MW/Fsp3 space [FDA-approved drugs (dark gray), spiro-bis-morpholine library compounds (orange)].
Six-membered cyclic amines (piperidines, piperazines, and morpholines) abound in bioactive compounds. By contrast, seven-membered cyclic amines are far less common,^43^ and their incorporation into 6,7-spirocycles is even more unusual;^3b,10a,12a^ thus, the 6,7-spirocyclic homologue of 6,6-spiroacetal 8 would represent a novel scaffold for compound library assembly. Iodoacetalization of enol ether 4 using N-Boc-propanolamine proceeded uneventfully to provide the corresponding iodide. Subsequent formation of the 1,4-oxazepane using our established cyclization conditions proved inefficient; the slow reaction led to competing formation of a urea byproduct 18 (Scheme 4).^44^ The base-mediated elimination of t-BuOH from Boc carbamates to afford an isocyanate has been reported.^45^ We postulate urea 18 arises from isocyanate trapping with dimethylamine, which originates from the slow decomposition of the DMF solvent under the reaction conditions.^46^ Urea formation was suppressed by performing the reaction at higher concentration (0.2 M) and using 2 equiv of t-BuOK; the increased rate of cyclization now allowed isolation of oxazepane 19 in 70% yield. Exchanging the benzyl protecting group in 19 for a Cbz carbamate provided orthogonally protected 6,7-spiroacetal 20 in a high yield (Scheme 4). Efforts to synthesize the corresponding 6,8- and 6,9-spiroacetals failed (see the Supporting Information).
Formation of Spiroacetals Incorporating 1,4-Oxazepanes
As a complementary strategy, 2-methylidene-1,4-oxazepane 21 was synthesized from 3-benzylaminopropan-1-ol and epichlorohydrin, using our established method (see the Supporting Information). Iodoacetalization with N-Boc-ethanolamine (5), followed by morpholine ring formation, provided 7,6-spiroacetal 22 in 46% yield over two steps. Exchanging the benzyl protecting group in 22 for a Cbz carbamate provided the orthogonally protected 7,6-spiroacetal scaffold 23. Crystals suitable for analysis by single-crystal X-ray diffraction confirmed the structure of 23, with the spiroacetal benefiting from double anomeric stabilization (see the Supporting Information). As a final example, bis-oxazepane spiroacetal 24 was also synthesized via this route. While the formation of the intermediate iodoacetal proceeded in good yield from enol ether 21 and N-Boc-propanolamine, subsequent cyclization to install the second oxazepane ring furnished 24 in low yield. No efforts were made to optimize this reaction. 6,7-Spiroacetal 20 was accessed on >50 g scale.^47^ Hydrogenolysis of the Cbz carbamate afforded secondary amine 25 in good yield. Amine functionalization with a representative amide (26), sulfonamide (27) and urea (28), followed by Boc deprotection, provided 6,7-spiroacetals 29,^48^30·2HCl, and 31·HCl, respectively (Scheme 5), from which a small library of 33 compounds was synthesized without event (see the Supporting Information).^49^ This small study suggests that spiroacetal scaffolds containing 1,4-oxazepanes are viable starting materials for compound library assembly.
Orthogonal Deprotection and Functionalization of 6,7-Spiroacetal Scaffold 20Reaction conditions: 4-methyloxazole-5-carboxylic acid, HATU, iPr2NEt, CH2Cl2, 0 °C–rt, 16 h, 79% (26); or pyridine-3-sulfonyl chloride, Et3N, CH2Cl2, 0 °C–rt, 16 h, 89% (27); or 2,4-difluoro-1-isocyanatobenzene, Et3N, CH2Cl2, rt, 2 h, 89% (28).
The introduction of even a methyl substituent^50,51^ can drastically affect lipophilicity, solubility, bioavailability, and selectivity of bioactive compounds;^52^ thus, being able to access methylated and similarly substituted spiroacetal scaffolds would potentially be useful for future hit optimization studies. 2-Chloromethyl-6-methylmorpholine 32 was produced in good yield under our standard conditions from (S)-1-benzylaminopropan-2-ol (33) and rac-epichlorohydrin (3) and isolated as a ∼2:1 mixture of diastereoisomers^53^ (Scheme 6). Dehydrochlorination provided enol ether 34 in 71% yield.^54^ Incorporating a methyl substituent at the 5-position of the morpholine proved more challenging: analogous reaction of (S)-2-benzylaminopropan-1-ol (35) with rac-epichlorohydrin (3) afforded 6-regioisomer 32, again as a ∼2:1 mixture of diastereomers; the desired 2,5-disubstituted regioisomer 36 was not observed. LCMS analysis of the first step in both reactions revealed the formation of a 1:1 mixture of two chlorohydrin diastereoisomers (37 from 33 and 38 from 35), consistent with both enantiomerically pure aminoalcohols reacting similarly with the racemic epoxide. This observation suggested that the formation of the 2,6-regioisomer from 35 was occurring in the second step of the reaction. Performing this step at different temperatures (150, 170, and 190 °C) had no impact on the outcome; however, the stoichiometry of acid did: using 1.0 equiv of 98% H_2_SO_4_ led to exclusive formation of 2,6-disubstituted regioisomer 32; using 2.0 equiv of acid, both morpholine regioisomers were observed in a 2:1 ratio, with the 2,6-regioisomer now the minor product. Formation of this regioisomer was suppressed completely when 4.0 (or greater) equiv of acid was employed;^55,56^ under these conditions, morpholine 36 was obtained in 62% yield (Scheme 6).^54^ In the presence of excess acid, we tentatively propose that the amine in 38 is effectively permanently protonated, suppressing the formation of aziridinium species,^57^ which provide a pathway for isomerization (see the Supporting Information).
Formation of Methyl-Substituted Bis-morpholine Spiroacetals 42 and 43
Dehydrohalogenation of regioisomers 32 and 36 provided 2-methylidenemorpholines 34 and 39, respectively, in good yields (Scheme 6). Iodoacetalization proceeded efficiently on both enol ethers. No stereoinduction was observed in the reaction of 5-methyl-substituted enol ether 39; the iodoacetal product 40 was isolated as a 1:1 mixture of diastereoisomers. Moving the methyl substituent to the 6-position of the morpholine ring (34) led to modest diastereoselectivity, with iodoacetal 41 now formed as a 4:1 mixture of diastereoisomers. In both cases, the diastereoisomeric ratio did not change when the reaction was performed at 0 or −78 °C. Finally, cyclization to introduce the second morpholine ring afforded 5- and 6-methyl-substituted spiroacetals, 42 and 43, respectively (Scheme 6). In both cases, the diastereoisomeric ratio of the iodoacetal starting materials was preserved in the spiroacetal products, suggesting efforts to improve the diastereomeric ratio, for example, using a chiral electrophilic iodine reagent would be worth exploring in the future. It was not possible to determine by NMR spectroscopy the orientation of the methyl substituent in the two diastereoisomers of spiroacetal 42 (5-Me) owing to resonance overlap and the presence of rotamers; however, the appearance of the resonance for one of the C(5)Hs in the major diastereoisomer of spiroacetal 43 (6-Me) as an apparent triplet [δ_H_ = 1.81 (app t, J = 10.9 Hz)] is consistent with an axially oriented hydrogen substituent at C(6) (Scheme 6). We were unable to confirm the stereochemistry of the spiroacetal; however, based on the crystal structure of spiroacetal 7 (Figure 2) and literature precedent,^58^ we hypothesize that this diastereoisomer is the double anomerically stabilized spiroacetal with an equatorial methyl substituent.
Incorporating substituents into the ring formed in the final cyclization step proved to be more straightforward. Ten commercially available, enantiopure (where applicable) Boc-protected aminoalcohols were used to produce substituted spiroacetals 44–54 (Figure 4). In most cases, the spiroacetals were isolated in greater than 60% yield from 2-methylidenemorpholine 4 and, where relevant, as 1:1 mixtures of diastereoisomers, highlighting that the stereochemical information embedded in the nucleophile imparted no stereoinduction. The formation of 1,1-dimethyl-2-isopropyl-substituted spiroacetal 51 was low-yielding, presumably because of the sterically hindered tertiary alcohol employed in the iodoacetalization step. The reaction sequence to form benzene-fused spiroacetal 53 was also low-yielding, which is likely a consequence of the lower nucleophilicity of both the phenol in the iodoacetalization step and the aniline carbamate in the cyclization. The highest overall yield was observed for the formation of gem-dimethyl-substituted spiroacetal 49, which may be attributed to the Thorpe–Ingold effect facilitating ring closure.^59^
Synthesized substituted bis-morpholine spiroacetals. aSynthesized from enol ether 39.
Racemates are commonly employed in high-throughput screens of compound libraries; however, the inclusion of diastereoisomeric mixtures is generally avoided. Lewis acids have been used to epimerize heterocyclic acetals;^60^ however, we focused on the use of Brønsted acids, pioneered by Deslongchamps on similarly substituted 6,6-spiroacetals (1,7-dioxaspiro[5.5]undecanes), to explore the possibility of improving the diastereoisomeric ratio of substituted bis-morpholine spiroacetals by anomerization.^58^ Unsurprisingly, the attempted anomerization of spiroacetal 48 using 3 M HCl in MeOH led to Boc deprotection. An analogue (55) containing Cbz and Fmoc carbamate protecting groups was therefore synthesized (see the Supporting Information). A 1:1 diastereoisomeric mixture of 55 was treated with 3 M HCl in MeOH. After 24 h at room temperature, the ratio had increased to 9:1, as determined by LCMS.^61^ This ratio did not change over the course of 1 week, suggesting the reaction had reached equilibrium. It was not possible to confirm the structure of the two diastereoisomers by ^1^H NMR spectroscopy owing to extensive resonance overlap, compounded by the presence of rotamers; however, based on the crystal structure of spiroacetal 7 (Figure 2) and literature precedent,^58^ we postulate that the major diastereoisomer is the double anomerically stabilized spiroacetal 55a, in which the benzyl group occupies an equatorial orientation (Scheme 7).^62^ Subjecting a 3:2 diastereoisomeric mixture of doubly substituted bis-morpholine spiroacetal 56 (see the Supporting Information) to 3 M HCl in MeOH at room temperature delivered a 19:1 (by LCMS) ratio of products after 22 h; this ratio did not change upon extension of the reaction time. We again hypothesize the major diastereoisomer is 56a, which benefits from double anomeric stabilization and now two substituents adopting equatorial positions;^63^ this result suggests that in some instances (especially matched cases), it should be possible to access highly diastereoisomerically enriched products by anomerization.
Anomerization of Spiroacetals 55 and 56
Conclusions
Morpholines are ubiquitous in drug discovery. In this study, we report a four-step synthesis of a scaffold containing two such heterocycles embedded within a spiroacetal framework. The synthesis involves the intermediacy of a 2-chloromethyl-substituted morpholine, which undergoes elimination to afford an exocyclic enol ether, from which the second morpholine ring is constructed in two steps. The overall synthesis is high-yielding and can be performed on a large scale from readily available starting materials. The method can be extended to the generation of 6,7- and 7,7-spiroacetal analogues and to substituted 6,6-systems. Substitution introduces issues of diastereoselectivity, which in some instances can be improved by acid-mediated anomerization. The two amine functionalities embedded in the 6,6- and 6,7-spiroacetal scaffolds can be sequentially functionalized, providing entry into compound libraries that occupy drug-like chemical space. These spiroacetals are hydrolytically stable and, therefore, represent attractive starting materials for drug discovery.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Stockdale T. P.; Williams C. M. Pharmaceuticals that Contain Polycyclic Hydrocarbon Scaffolds. Chem. Soc. Rev. 2015, 44, 7737–7763. 10.1039/C 4CS 00477 A.26171466 · doi ↗ · pubmed ↗
- 2Chupakhin E.; Babich O.; Prosekov A.; Asyakina L.; Krasavin M. Spirocyclic Motifs in Natural Products. Molecules 2019, 24, 416510.3390/molecules 24224165.31744211 PMC 6891393 · doi ↗ · pubmed ↗
- 3a Hiesinger K.; Dar’In D.; Proschak E.; Krasavin M. Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183. 10.1021/acs.jmedchem.0c 01473.33381970 · doi ↗ · pubmed ↗
- 4a Bagdanoff J. T.; Chen Z.; Acker M.; Chen Y. N.; Chan H.; Dore M.; Firestone B.; Fodor M.; Fortanet J.; Hentemann M.; Kato M.; Koenig R.; Labonte L. R.; Liu S.; Mohseni M.; Ntaganda R.; Sarver P.; Smith T.; Sendzik M.; Stams T.; Spence S.; Towler C.; Wang H.; Wang P.; Williams S. L.; Lamarche M. J. Optimization of Fused Bicyclic Allosteric SHP 2 Inhibitors. J. Med. Chem. 2019, 62, 1781–1792. 10.1021/acs.jmedchem.8b 01725.30688462 · doi ↗ · pubmed ↗
- 5a Degorce S. L.; Bodnarchuk M. S.; Scott J. S. Lowering Lipophilicity by Adding Carbon: Aza Spiro Heptanes, a log D Lowering Twist. ACS Med. Chem. Lett. 2019, 10, 1198–1204. 10.1021/acsmedchemlett.9b 00248.31417667 PMC 6693472 · doi ↗ · pubmed ↗
- 6a King T. A.; Stewart H. L.; Mortensen K. T.; North A. J. P.; Sore H. F.; Spring D. R. Cycloaddition Strategies for the Synthesis of Diverse Heterocyclic Spirocycles for Fragment-Based Drug Discovery. Eur. J. Org. Chem. 2019, 5219–5229. 10.1002/ejoc.201900847.PMC 677428731598091 · doi ↗ · pubmed ↗
- 7Stotani S.; Lorenz C.; Winkler M.; Medda F.; Picazo E.; Ortega Martinez R.; Karawajczyk A.; Sanchez-Quesada J.; Giordanetto F. Design and Synthesis of Fsp 3-Rich, Bis-Spirocyclic-based Compound Libraries for Biological Screening. ACS Comb. Sci. 2016, 18, 330–336. 10.1021/acscombsci.6b 00005.27163646 · doi ↗ · pubmed ↗
- 8Perron F.; Albizati K. F. Chemistry of Spiroketals. Chem. Rev. 1989, 89, 1617–1661. 10.1021/cr 00097 a 015. · doi ↗