Identification and removal of a cryptic impurity in pomalidomide-PEG based PROTAC
Bingnan Wang, Yong Lu, Chuo Chen

TL;DR
This paper identifies and addresses a hidden impurity formed during the synthesis of a drug used for targeted protein degradation.
Contribution
The study reveals a previously overlooked byproduct in PROTAC synthesis and proposes a simple method to remove it.
Findings
A nucleophilic acyl substitution byproduct co-elutes with the desired product during HPLC.
Taurine scavenging effectively reduces contamination from the byproduct.
The impurity is generated due to competing reactions during the synthesis process.
Abstract
Chemically induced dimerization is a powerful tool for studying protein function, wherein the IMiD (the “immunomodulatory drug”) class of PROTAC molecules with a PEG linker is frequently used to promote targeted protein degradation. The standard protocol for their synthesis involves nucleophilic aromatic substitution of 4-fluorothalidomide with a PEG-amine. We report herein the identification of a commonly ignored impurity generated in this process. Nucleophilic acyl substitution competes with aromatic substitution to displace glutarimide and gives a byproduct that can co-elute with the desired product on HPLC throughout the remainder of the synthesis. Scavenging with taurine is a convenient way to minimize this contamination.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
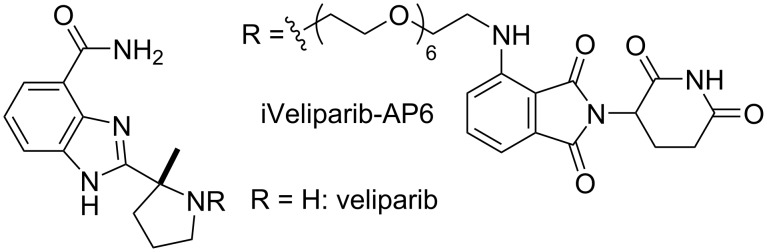 Figure 1
Figure 1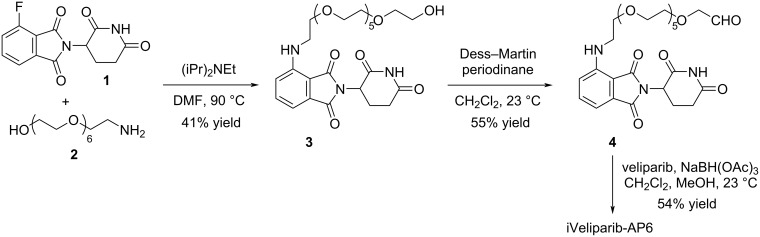 Scheme 1
Scheme 1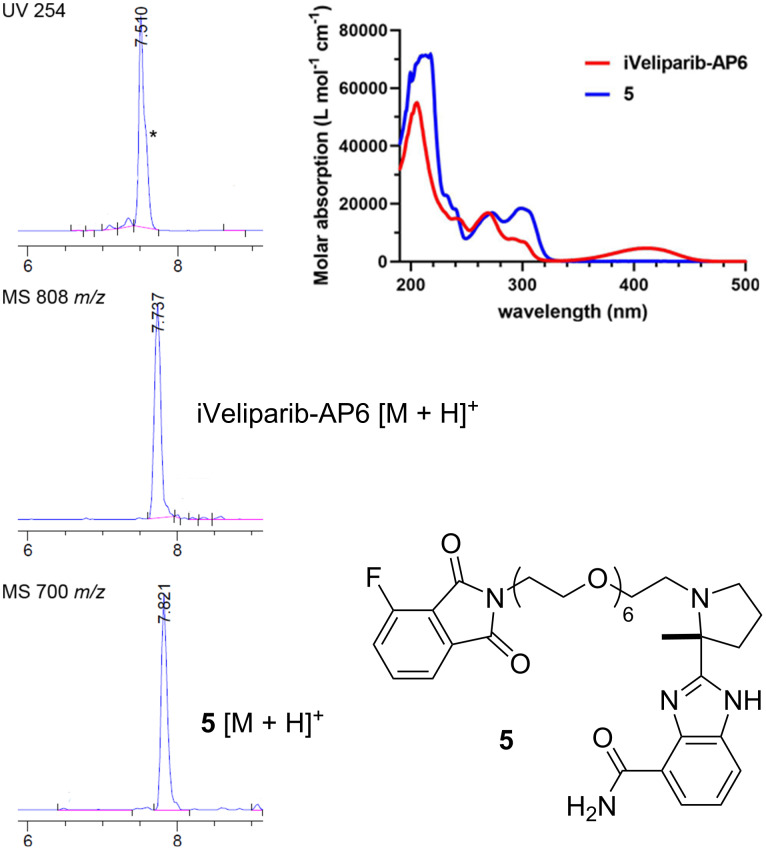 Figure 2
Figure 2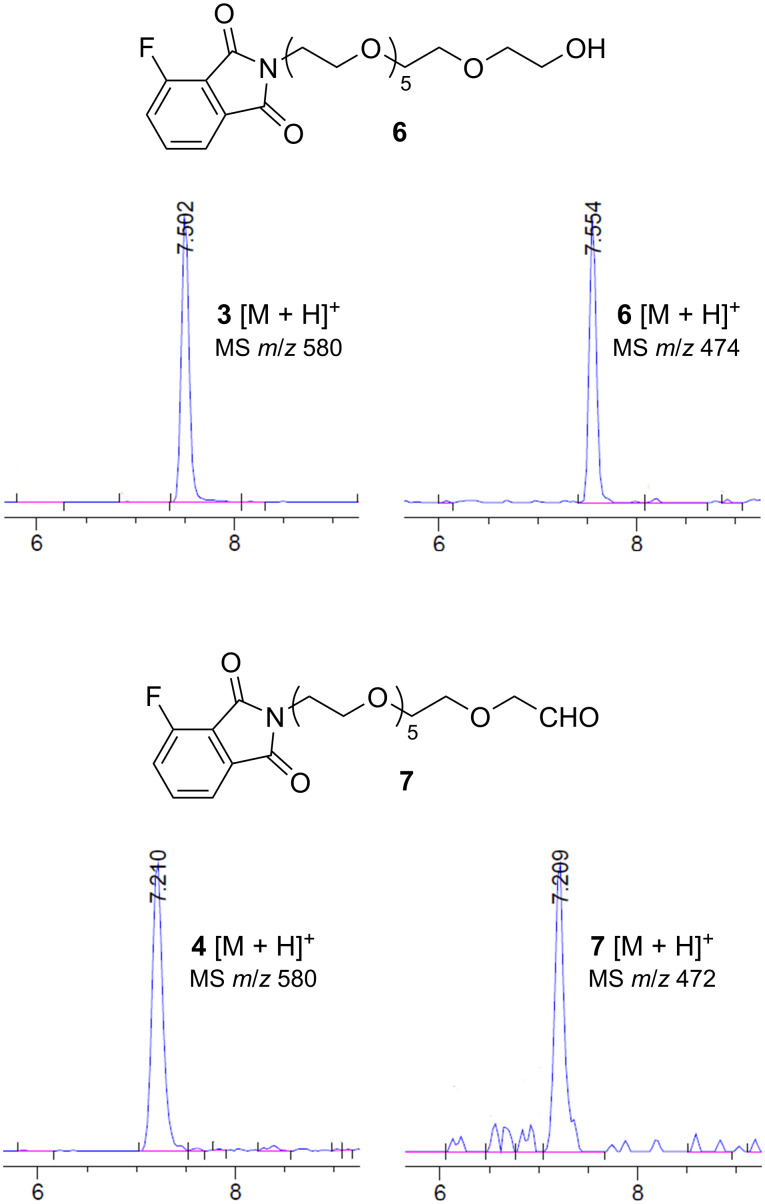 Figure 3
Figure 3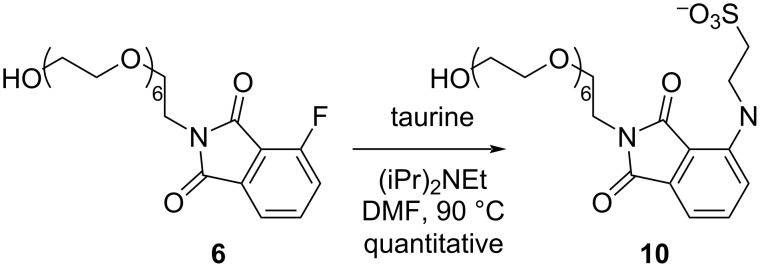 Scheme 2
Scheme 2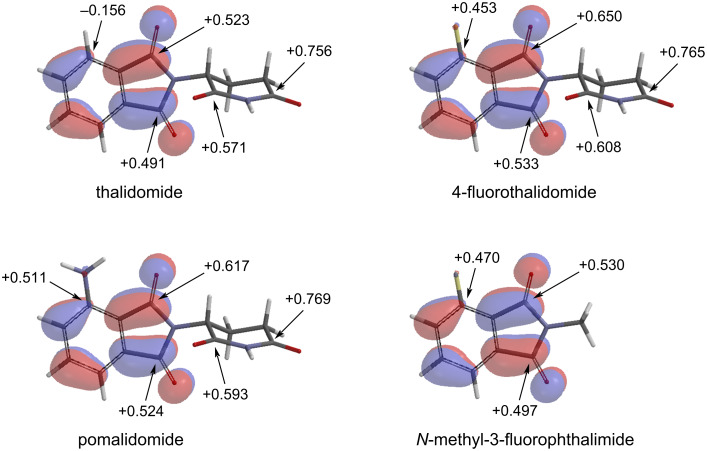 Figure 4
Figure 4|
| ||
|
| ||
| Entry | R | Δ (rt |
|
| ||
| 1 |
| 0.1 min |
| 2 |
| <0.1 min |
| 3 |
| <0.1 min |
| 4 |
| <0.1 min |
| 5 |
| <0.1 min |
| 6 |
| <0.1 min |
| 7 |
| 0.2 min |
| 8 |
| 0.5 min |
| 9 |
| 0.1 min |
| 10 |
| 0.5 min |
| 11 |
| 0.3 min |
- —UTSW SCCC
- —National Cancer Institutehttps://doi.org/10.13039/100000054
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProtein Degradation and Inhibitors · Multiple Myeloma Research and Treatments · Ubiquitin and proteasome pathways
Introduction
Targeted protein degradation capitalizing on the concept of chemically induced dimerization has emerged as a new therapeutic approach recently [1]. In particular, the modularity of proteolysis targeting chimera (PROTAC) has made it a popular starting point to develop selective small-molecule degraders [2]. Currently, leveraging ubiquitination by the von Hippel–Lindau (VHL) protein or cereblon (CRBN) is the most successful method to achieve targeted protein degradation [3–4]. For initial studies, a short polyethylene glycol (PEG) linker of various lengths is typically used to build a small library to identify a lead compound. For example, iVeliparib-AP6 (Figure 1) developed through this practice is a PROTAC that degrades poly(ADP-ribose) polymerase 2 (PARP2) selectively [5].
The structures of veliparib and iVeliparib-AP6.
Results and Discussion
The synthesis of iVeliparib-AP6 [5] starts with a nucleophilic aromatic substitution (S_N_Ar) reaction wherein 4-fluorothalidomide (1) reacts with amino-PEG7-OH 2 to give alcohol 3 (Scheme 1). Subsequent alcohol oxidation followed by reductive amination of the resulting aldehyde 4 with veliparib [6–7] provides iVeliparib-AP6. As a common practice for small-molecule library synthesis, the identity and the purity of the reaction products of this simple, 3-step process were analyzed by LC–MS.
The synthetic route for iVeliparib-AP6.
Whereas the HPLC traces of the reaction mixtures showed one major product peak, the final product carried a barely noticeable shoulder peak (Figure 2, marked with asterisk). A close examination of the UV profile indicated the presence of an impurity. Its removal requires repetitive prep-HPLC purifications that are inefficient and time-consuming. The isolated impurity lacks the characteristic fluorescent yellow color of pomalidomide or its close analogs, indicating that a side reaction unrelated to the S_N_Ar of the fluoride has occurred. ^1^H NMR and MS analyses suggested that phthalimide 5 was the byproduct formed through this series of transformations.
Identification of the cryptic impurity of iVeliparib. The UV trace of the initially purified iVeliparib-AP6 and the MS ion extraction traces of the desired product and the byproduct 5 on HPLC.
Supporting this hypothesis, analysis of the reaction intermediates confirmed the presence of 6 that similarly co-eluted with 3 and 7 that completely overlapped with 4 on HPLC under our standard conditions (MeCN/0.1% TFA in water, 10%→60% 0→7 min, 60%→100% 7→10 min, 100% 10→15 min) (Figure 3). The identity of these impurities was further confirmed by independent synthesis of 5 using the same sequence of reactions starting from 3-fluorophthalic anhydride instead of 1. Indeed, 5 is a white solid without UV absorption around 410 nm (Figure 2).
The structures of 6 and 7 and the MS ion extraction traces of 3, 4, 6, and 7 from HPLC analysis of the reaction mixtures after purification by silica gel column chromatography.
The formation of 6 originated from nucleophilic acyl substitution to displace the glutarimide in 1 by 2. Interestingly, this side reaction was concentration dependent, giving various amounts of 6 under otherwise the same reaction conditions. For example, at 10–30 mM wherein most of the pilot studies were conducted, the ratio of 3 and 6 could be as low as ≈1:50 by ^1^H NMR and ≈1:10 by MS analysis. Under these conditions, the formation of 6 can evade detection if unaware of this issue. However, the amount of 6 can increase to as much as 20% when conducting the reaction at 0.1–0.3 M, the concentrations generally used for preparative work. Notably, because the S_N_Ar reaction of 1 proceeded slowly, 4-(dimethylamino)thalidomide also formed due to the decomposition of DMF. Whereas switching the solvent could prevent its formation, the accumulation of 7 remained an issue, giving no substantial improvement in the yield of 3 in, for example, NMP. In MeCN, glutarimide displacement became a major reaction. Because NMP was significantly more difficult to remove after the reaction, and 4-(dimethylamino)thalidomide could be separated from 3 easily, we opted to keep DMF as the solvent. We only detected by LC–MS a trace amount of the secondary byproduct derived from S_N_Ar of 6 or glutarimide displacement of 3 by 2.
To understand the extent by which glutarimide displacement affects quality control analysis, we tested a series of amines and compared the retention times of 8 and 9 (Table 1). Rather surprisingly, the length of amino-PEG-OH had no effect on the relative retention time of 8 and 9. The byproduct co-eluted with the desired product on HPLC even for the reaction of diethylene glycolamine and 1. Capping the free hydroxy group with a methyl group improved the separation on HPLC marginally. However, incorporating a clickable propargyl group greatly benefited separation. Similar to the alcohol, amino-PEG5-acid also reacted with 1 to generate an inseparable byproduct. By contrast, the byproduct derived from Boc-protected amines has a significantly different retention time on HPLC and could be removed by regular silica gel chromatography.
Because the formation of 5‒7 posts a significant purification challenge, we sought to develop a method to facilitate the elimination of this impurity. We first attempted scavenging 6 by solid-phase supported amines. Incubating a mixture of 3 and 6 with TentaGel S-NH_2_ in DMF led to a gradual decrease of 6 over four days. Whereas this method is applicable to removing 9 of different PEG-OH length (including 6 for n = 5), the decontamination process is slow and the costs are high. Additionally, it could not remove the trace amount of the residual impurity, potentially due to inefficient diffusion of the substrate into the resin at low concentrations. We next sought to use soluble amines to enable an easy separation of the byproduct. Pleasingly, reacting the mixture of 3 and 6 with taurine converted 6 to sulfonate 10 (Scheme 2) that can be removed by a simple aqueous wash with sodium bicarbonate. Whereas taurine may also react with 3, the overall yield of 3 was not affected significantly. In a typical run, we isolated a 3:1 mixture of 3 and 6 in 54% combined yield and obtained pure 3 in 41% yield after taurine treatment. We did not observe any byproduct corresponding to glutarimide displacement of 3 by taurine. Using this method, we could easily prepare 100 mg of iVeliparib-AP6 with <1% impurity based on ^1^H NMR and MS analysis.
The conversion of byproduct 6 to 10 to facilitate its removal.
The mechanism by which glutarimide displacement competes with S_N_Ar merits some discussion. Thalidomide is well known for its configurational instability and racemizes rapidly [8]. However, equally important but much less appreciated is its hydrolytic instability, where the cleavage of the phthalimide and glutarimide rings comprises the major metabolic pathways [9–10]. Computational analysis confirms that C1, C3, C2’, and C4’ are the most electrophilic sites of thalidomide (Figure 4). As expected, introducing a fluorine atom to C4 activates it toward S_N_Ar. Although C2’ and C4’ remain the most electropositive sites, we could not isolate any byproducts corresponding to reactions with glutarimide. The preferred nucleophilic reaction at C1, C3, and C4 can be explained by the fact that the LUMO of 1 resides entirely on phthalimide. The low S_N_Ar reaction rate may also be explained by C4 being located near the node of the LUMO. However, S_N_Ar is still favored over glutarimide displacement potentially because the negative charge in the corresponding intermediate is stabilized by an extended conjugation system. In contrast, the negative charge in the carbonyl addition intermediate is stabilized by an oxygen atom only. As such, the erosion of 3 by taurine was minimal.
The LUMO and electrostatic charges of thalidomide and its derivatives by DFT calculation at the ωB97X-D/6-311+G* level.*
Conclusion
Nucleophilic aromatic substitution of 4-fluorothalidomide (1) has provided a convenient entry to the IMiD class of PROTAC molecules. Although the yield of the desired product is generally modest, it remains highly popular because of the convenience and the modularity of this method. When reacting 1 with an amino-PEG-OH, the purification of the reaction mixture is particularly difficult as a major byproduct co-elutes with the desired product even on HPLC. Importantly, the impurity issue can persist throughout the remainder of the synthesis if not addressed directly. To our knowledge, there has been no discussion regarding the identity of this impurity. We show herein that nucleophilic acyl substitution competes with aromatic substitution to displace the glutarimide. Introducing a sulfonate to the byproduct by reacting it with taurine allows for easy decontamination. This method may facilitate the purification of other similar reactions and improve the quality of related pomalidomide derivatives.
Supporting Information
File 1Computational details, general experimental information, synthetic procedures, compound characterization data, and copies of NMR spectra.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Schreiber S L Cell 20211843910.1016/j.cell.2020.12.02033417864 · doi ↗ · pubmed ↗
- 2Chirnomas D Hornberger K R Crews C M Nat Rev Clin Oncol 20232026527810.1038/s 41571-023-00736-336781982 PMC 11698446 · doi ↗ · pubmed ↗
- 3Sakamoto K M Kim K B Kumagai A Mercurio F Crews C M Deshaies R J Proc Natl Acad Sci U S A 2001988554855910.1073/pnas.14123079811438690 PMC 37474 · doi ↗ · pubmed ↗
- 4Winter G E Buckley D L Paulk J Roberts J M Souza A Dhe-Paganon S Bradner J E Science 20153481376138110.1126/science.aab 143325999370 PMC 4937790 · doi ↗ · pubmed ↗
- 5Wang S Han L Han J Li P Ding Q Zhang Q-J Liu Z-P Chen C Yu Y Nat Chem Biol 2019151223123110.1038/s 41589-019-0379-231659317 PMC 6864272 · doi ↗ · pubmed ↗
- 6Penning T D Zhu G-D Gandhi V B Gong J Liu X Shi Y Klinghofer V Johnson E F Donawho C K Frost D J J Med Chem 200952251452310.1021/jm 801171 j 19143569 · doi ↗ · pubmed ↗
- 7Kolaczkowski L Barkalow J Barnes D M Haight A Pritts W Schellinger A J Org Chem 2019844837484510.1021/acs.joc.8b 0286630716275 · doi ↗ · pubmed ↗
- 8Yamamoto T Tokunaga E Nakamura S Shibata N Toru T Chem Pharm Bull 20105811011210.1248/cpb.58.11020045977 · doi ↗ · pubmed ↗