Dihydrolipoamide dehydrogenase deficiency in two unrelated Tunisian children
Hajer Aloulou, Fatma Charfi, Rim Charfi, Amel Ben Chehida, Hela Boudabbous, Imen Chabchoub, Elise Lebigot, Thouraya Kammoun, Ines Maaloul

TL;DR
Two Tunisian children with a rare metabolic disorder called dihydrolipoamide dehydrogenase deficiency are described, highlighting the condition's varied symptoms and the importance of genetic testing for diagnosis.
Contribution
This is the first report of dihydrolipoamide dehydrogenase deficiency in Tunisia, providing new clinical and genetic insights into the disease's phenotypic variability.
Findings
DLDD was confirmed in two unrelated Tunisian children through Sanger sequencing of the DLD gene.
The pathogenic variant c.685G > T (p.Gly229Cys) was found in a homozygous state in both patients.
The disease's biochemical markers are variable, emphasizing the need for genetic confirmation.
Abstract
Dihydrolipoamide dehydrogenase deficiency (DLDD) (OMIM# 246,900) is an extremely rare inherited metabolic disorder causing neurological and/or liver impairment. The clinical manifestations are mostly characterized by severe neurological impairment in early childhood, hepatic presentations and rarely by myopathic manifestations. Here, we describe two patients presenting with recurrent episodes of vomiting and liver dysfunction. DLDD was confirmed via sanger sequencing by identification of the pathogenic variant c.685G > T (p.Gly229Cys) in DLD gene at a homozygous state. To our knowledge, this is the first Tunisian report of DLDD. Phenotypic spectrum of this disease is very large. Biochemical markers that predict the impairment of the pathways affected by the deficiency of E3 subunit (gluconeogenesis, tricyclic cycle and catabolism of branched chain aminoacids) are variably present.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
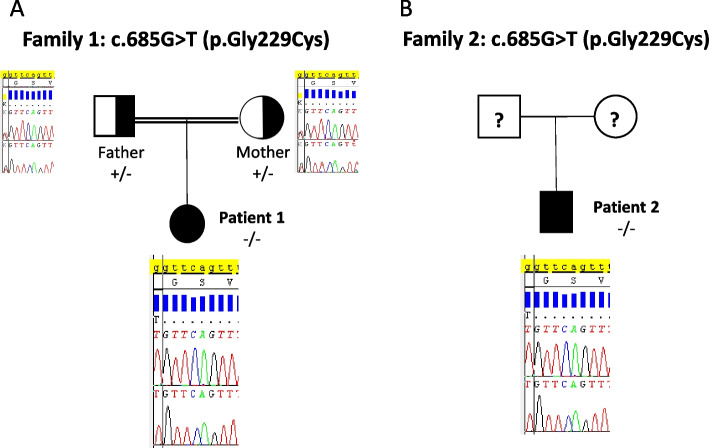 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBiochemical Acid Research Studies · Metabolism and Genetic Disorders · Biochemical and Molecular Research