Remodeling of extracellular matrix collagen IV by MIG-6/papilin regulates neuronal architecture
Malika NADOUR, Robert I. VALETTE REVENO LEATIS, Marie BIARD, Noémie FRÉBAULT, Lise RIVOLLET, Philippe ST-LOUIS, Cassandra R. BLANCHETTE, Andrea THACKERAY, Paola PERRAT, Carlo BEVILACQUA, Robert PREVEDEL, Laurent CAPPADOCIA, Georgia RAPTI, Maria DOITSIDOU, Claire Y. BÉNARD

TL;DR
This study reveals how the protein MIG-6/papilin helps maintain the structure of neurons over a lifetime by remodeling collagen in the brain's extracellular matrix.
Contribution
The study identifies MIG-6/papilin as a novel regulator of neuronal architecture through extracellular matrix remodeling.
Findings
MIG-6/papilin remodels collagen IV in the extracellular matrix to maintain neuronal stability.
Collaboration with MIG-17/ADAMTS and PXN-2/peroxidasin is essential for this remodeling process.
The mechanism supports neuronal architecture under mechanical stress and over time.
Abstract
Neuronal architecture established embryonically must persist lifelong to ensure normal brain function. However, little is understood about the mechanisms behind the long-term maintenance of neuronal organization. To uncover maintenance mechanisms, we performed a suppressor screen in sax-7/L1CAM mutants, which exhibit progressive disorganization with age. We identified the conserved extracellular matrix protein MIG-6/papilin as a key regulator of neuronal maintenance. Combining incisive molecular genetics, structural predictions, in vivo quantitative imaging, and cutting-edge Brillouin microscopy, we show that MIG-6/papilin remodels extracellular matrix collagen IV, working in concert with the secreted enzymes MIG-17/ADAMTS and PXN-2/peroxidasin. This remodeling impacts tissue biomechanics and ensures neuronal stability, even under increased mechanical stress. Our findings highlight an…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
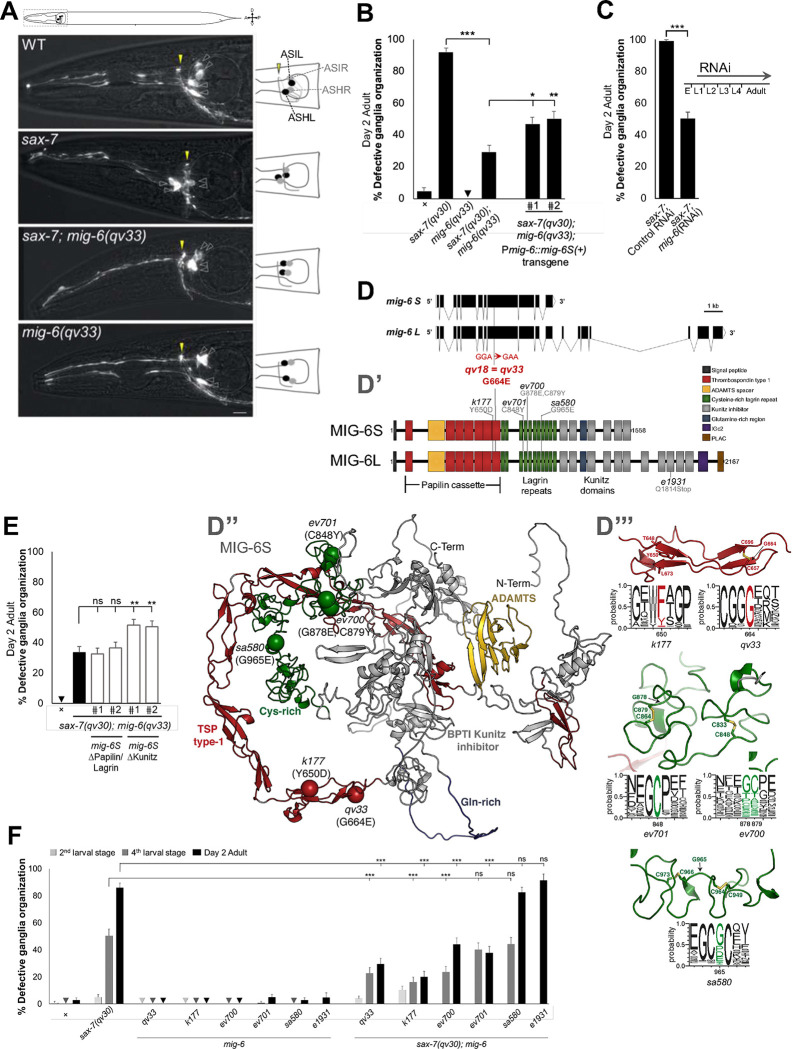 Figure 1
Figure 1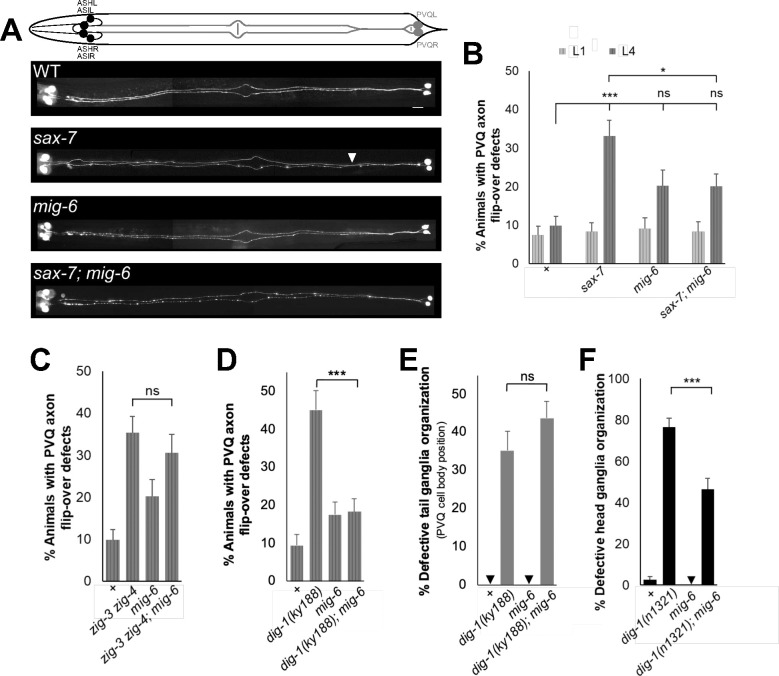 Figure 2
Figure 2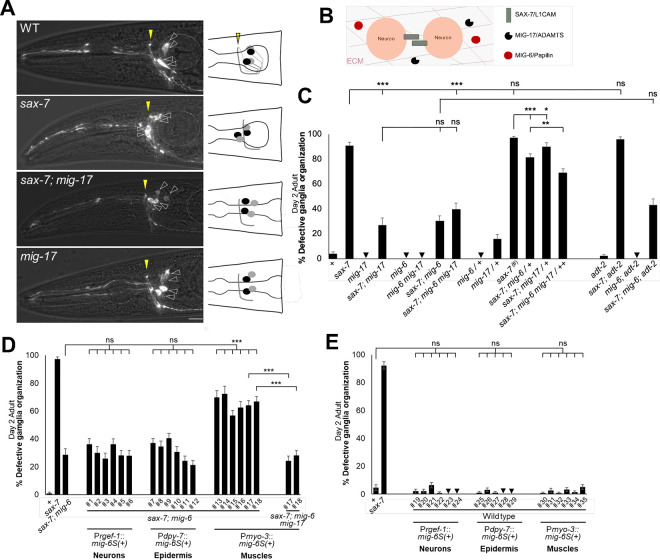 Figure 3
Figure 3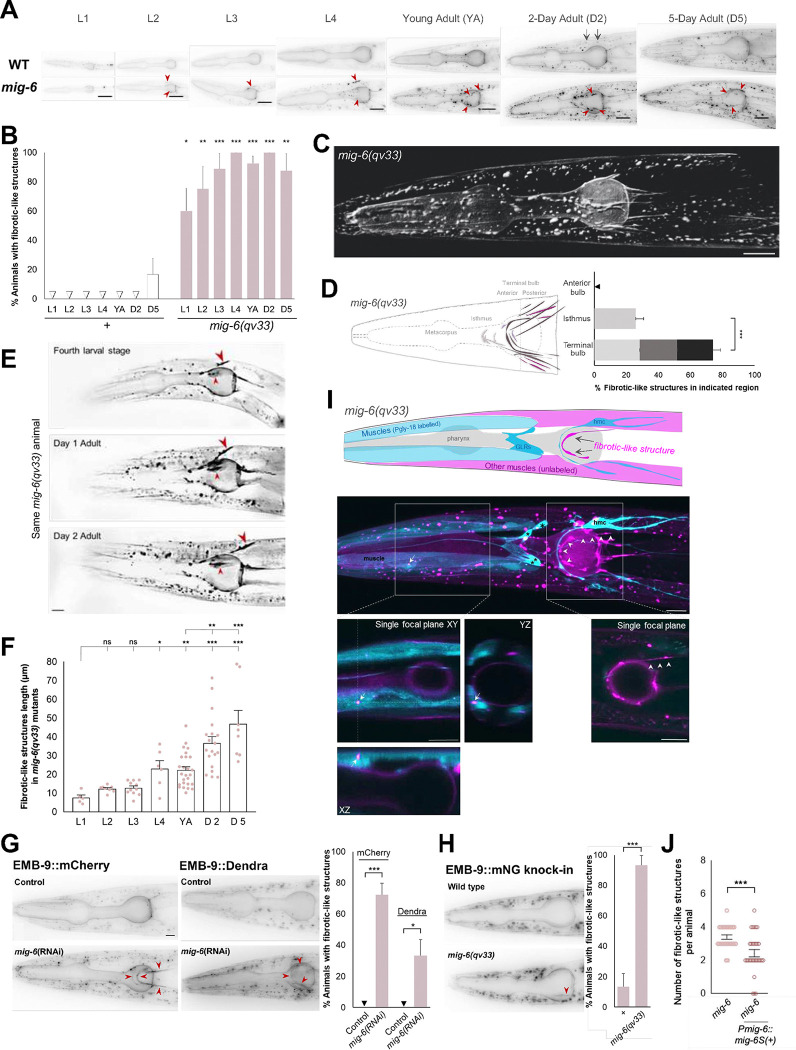 Figure 4
Figure 4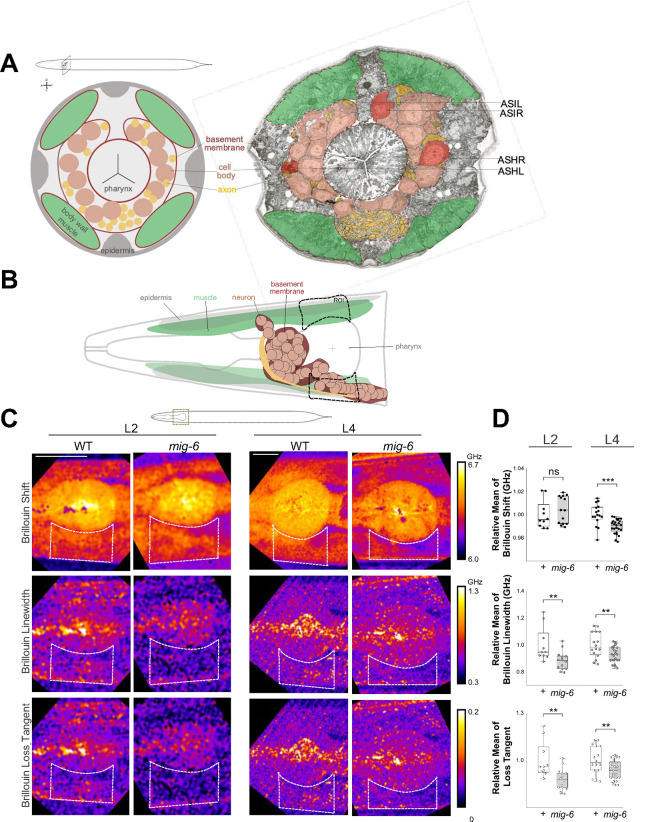 Figure 5
Figure 5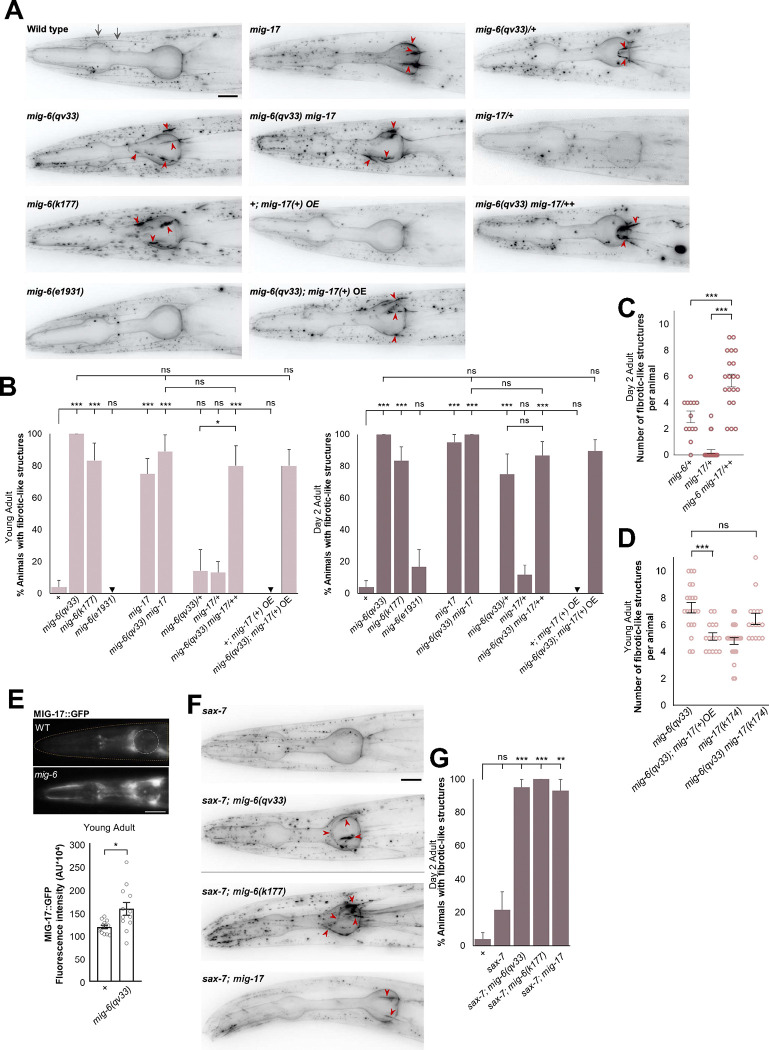 Figure 6
Figure 6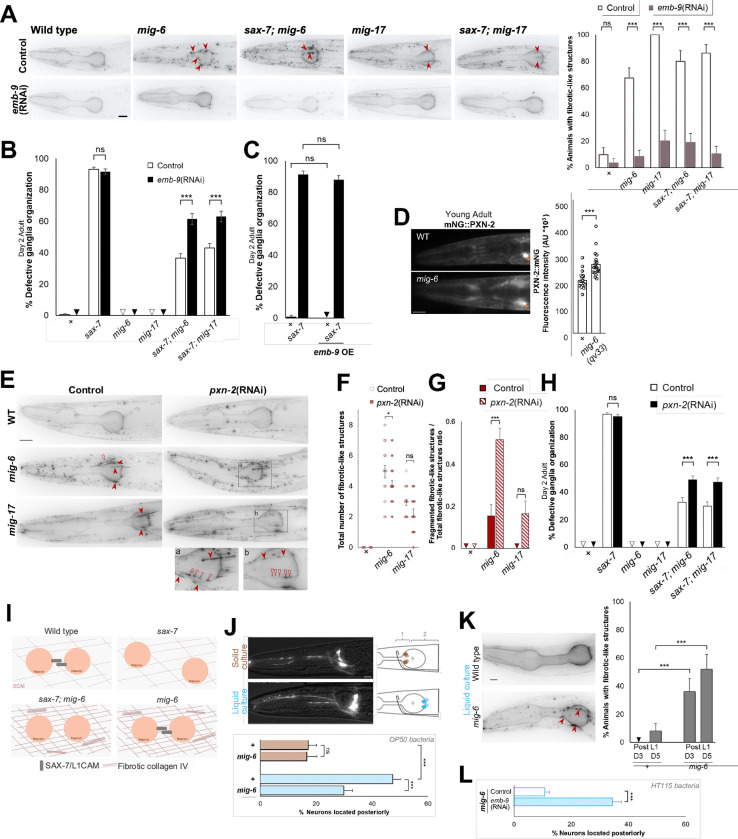 Figure 7
Figure 7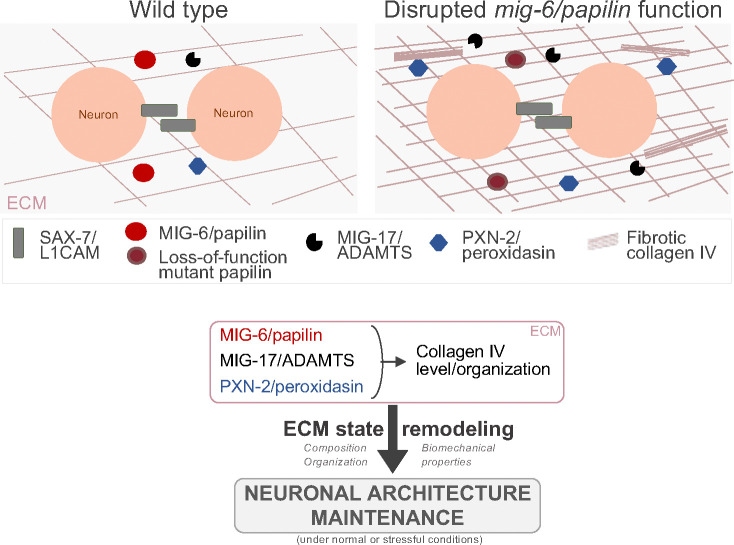 Figure 8
Figure 8- —National Science and Engineering Research Council of Canada
- —Fond de Recherche du Québec-Santé
- —Canadian Funds for Innovation-John Evan Leaders Equipment
- —Canadian Institutes of Health
- —National Institutes of Health of the USA
- —Fond de Recherche du Québec-Santé
- —CERMO-FC and UQAM M.Sc. and Ph.D. scholarships
- —CIHR scholarship
- —ERC Consolidator
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsConnective tissue disorders research · Cardiac Fibrosis and Remodeling · Macrophage Migration Inhibitory Factor
INTRODUCTION
Proper nervous system function depends on both developing and maintaining the intricate architecture of its neural circuits. The dynamic processes of development and maturation of the nervous system begin in utero and extend well into adulthood ^1^. Key neuronal features, established earlier in development, must be preserved throughout life to ensure continuity in neuronal architecture and function ^2^. The structural organization of the nervous system faces numerous challenges, including physical stresses induced by the organism’s postnatal growth, anatomical remodeling, and the integration of new neurons, body movements, and injury ^3^. Failure to stably maintain neuronal architecture over the long-term can impair and compromise neuronal function and contribute to the manifestation of neurological conditions ^4,5^. Notably, a defining feature of many neurodegenerative diseases is the destabilization of axons and dendrites, and accompanying loss of synapses ^6–8^. Gaining insights into the mechanisms that preserve nervous system architecture could inform the development of therapeutics aimed at preventing or reversing such neurological conditions. Despite their importance, the mechanisms that sustain neuronal organization throughout life remain poorly understood.
A key contribution to the preservation of the structural integrity of multicellular assemblies, including those in the nervous system, comes the extracellular matrix (ECM) ^9^. Cells interact with the ECM and neighboring cells via cell adhesion molecules, and the coordinated actions of these molecules and the ECM are key for cellular behavior, enabling, for instance, neuronal structures to withstand physical stresses ^5^. However, how cellular regulation implicating ECM interactions evolve through time, or when subjected to physical challenges, remains elusive.
The ECM is of crucial importance in the nervous system, playing critical roles during development ^10^, but also in the adult mammalian brain, where the ECM constitutes approximately 20% of its volume. Diffuse ECM is found near and between synapses across the brain, and condensed ECM is organized as basement membranes associated with blood vessels, or as lattice-like structures called perineuronal nets, surrounding the soma and dendrites of several neuron types in multiple brain regions ^11^. These organized forms of ECM influence neuronal biology in the adult brain, including dendritic spine stability, synapse plasticity, and axon regeneration ^11,12^. Also, changes in ECM composition and structure have been linked to various neurological diseases (e.g., Alzeihmer’s disease and schizophrenia), as well as to brain injury and aging ^13,14^. This underscores the critical role of the neurons’ extracellular environment in maintaining normal neuronal physiology and its involvement in pathological conditions ^5^. However, our understanding of the long-term regulation of the ECM in the mature nervous system is limited and remains a dauting problem for neuropathology ^15^. For instance, how the intricate interactions between ECM proteases and their substrates, regulate ECM dynamics for sustaining neuronal structure and function is poorly understood.
The nematode C. elegans provides a powerful in vivo genetic model to study the lifelong maintenance of nervous system architecture, and particularly the role of the ECM in this process. A significant number of C. elegans neurons are organized into ganglia, with most neuronal processes running along major fascicles, such as the neuropil and the ventral nerve cord ^16^. The multicellular assemblies of ganglia and nerve cords are ensheathed by a specialized ECM, namely basement membranes ^17^. After hatching into a larva, C. elegans undergoes a nearly 100-fold increase in size until it reaches adulthood ^18^. Yet, the overall architecture of its nervous system is established during embryogenesis and remains largely intact throughout post-embryonic growth ^17,19^. Indeed, serial-section electron microscopy and connectome reconstruction of multiple C. elegans brains at successive stages of development revealed that the shape and positioning of most neurons and neurites established at birth remains consistent through adulthood, including approximately 70% of adult brain synapses being part of stable connections that are proportionally maintained from birth to adulthood ^19^. Individual neurons and ECM components can be readily visualized in living animals throughout their lifespan, using fluorescent reporters thanks to C. elegans transparency and small size ^20,21^. Combined with its genetic tractability, including using cell-specific promoters and conditional knockdowns, these features enable the investigation of the mechanisms sustaining nervous system architecture across its lifetime.
Thus, in vivo genetic studies using C. elegans has yielded critical insights into the mechanisms of the long-term maintenance of neuronal assemblies. These investigations have revealed post-natal molecular mechanisms that actively preserve neuronal organization, particularly within ganglia and nerve cords. Notably, several immunoglobulin superfamily molecules play crucial roles in maintaining the architecture of these multi-neuronal assemblies over time. These include the cell adhesion molecule SAX-7/L1CAM ^22–27^, the large ECM protein DIG-1 ^28,29^, the secreted two-immunoglobulin domain containing proteins ZIG-3 and ZIG-4 ^30,31^, and the ectodomain of the FGF receptor EGL-15 ^32^. Mutations in the genes encoding these molecules lead to neuronal defects that arise later in development, well after the normal initial establishment of neuronal morphology of the affected neurons. Strikingly, neuronal maintenance defects are suppressed by paralysis, highlighting that neuronal structures experience internal mechanical stress generated by body or organ movements, and that the identified neuronal maintenance molecules counteract these stresses, which otherwise would lead to neuronal disorganization ^28,31,33^. Orthologues of some of these molecules have been found to sustain neural circuits in other systems as well; for instance, in mice, knockout of L1CAM specifically in the adult brain results in behavioral deficits and synaptic transmission changes ^34^, and L1CAM maintains neocortical axo-axonic innervation into adulthood ^35^. While we have evidence that cell adhesion molecule SAX-7/L1CAM and ECM molecule DIG-1 play important roles in maintaining nervous system architecture, how the underlying ECM landscape and molecular interactions contribute to sustaining nervous system architecture throughout life is not understood. Given the extensive evolutionary conservation of ECM and neuronal cell surface molecules from worms to mammals, the maintenance mechanisms unravelled in C. elegans will provide insights on general principles by which the nervous system architecture is preserved lifelong.
To expand our understanding of the molecular mechanisms governing the preservation of neuronal architecture throughout life, we conducted a forward genetic suppressor screen in the sax-7 mutant background, and identified mig-6, which encodes papilin, an extracellular matrix protein with structural similarities with ADAMTS metalloproteases ^36^. Papilin is conserved across metazoans, but its function remains elusive. Our findings reveal that MIG-6S/papilin is required post-developmentally for neuronal maintenance, by modulating the major extracellular matrix component collagen IV. Using Brillouin microscopy, we find that loss of mig-6 changes the biomechanical properties of tissues in the region comprising the neurons. We demonstrate that MIG-6S/papilin cooperates with the ECM remodeling metalloproteinase MIG-17/ADAMTS in regulating the collagen IV network, and that both collagen IV levels and crosslinking are critical to sustain neuronal organization lifelong. Our study underscores the critical role of ECM regulation by the extracellular matrix protein papilin in preserving neuronal architecture throughout an organism’s lifetime, including under conditions of significant mechanical stress. We propose a model in which a balance between flexibility and adhesion, mediated by ECM remodeling and cell adhesion, ensures the structural stability of the embryonically established nervous system over time and improve its ability to withstand mechanical stress.
MATERIALS AND METHODS
Please see Supplementary Information.
Data Availability Statement:
All data is available in the main text or the supplementary materials.
RESULTS
Loss of function of mig-6, which encodes the conserved extracellular matrix protein papilin, suppresses neuronal maintenance defects of sax-7 mutants
To identify novel genes involved in the long-term maintenance of neuronal organization, we conducted a forward genetic screen. We refrained from searching directly for mutants with neuronal maintenance defects, as previous efforts using this approach invariably yielded numerous alleles of the large neuronal maintenance gene dig-1 (^28^; C.Y.B., unpub. results). Rather, we reasoned that screening for suppressors of the defects of previously known neuronal maintenance mutants (Fig. S1), sax-7, would identify genes that directly or indirectly counteract defective long-term maintenance of neuronal architecture, providing insights into the basis of this process. In wild-type animals, the soma of chemosensory neurons ASH and ASI are located posterior to the nerve ring, where their axons project (neurons visualized with reporter P*sra-6::*DsRed2, Fig. 1A)^27^. This stereotypical positioning acquired during embryogenesis is preserved throughout life, making it a reliable indicator of neuronal organization. In sax-7 mutants, although the soma and axons of ASH/ASI initially exhibit normal positioning during earlier development, they later become displaced from the 4th larval stage onward (Fig. 1A,B,F) ^22,25,27^, with the ASH/ASI soma ending up anteriorly displaced, and the nerve ring shifting posteriorly, resulting in the soma aligning with or even anterior to the nerve ring (Fig. 1A). In our F2 clonal genetic screen for suppressors of sax-7 neuronal maintenance defects, we mutagenized sax-7 mutants with ethyl methanesulfonate and screened F3 broods by fluorescence microscopy to find suppressors of the sax-7 mutants ASH/ASI position defects. We isolated mutation qv18, which significantly suppressed the neuronal position defect in adult sax-7(qv24) animals, thus reducing the incidence of animals with mispositioned neurons (Fig. S1).
Through whole genome sequencing and bioinformatic analyses ^37–39^, we identified as a candidate mutation a G-to-A transition at transcript nucleotide 1991 of the gene mig-6, which causes a glycine to glutamic acid substitution at residue 664 (Fig. 1D’). To determine if this mutation was the causal suppressor, we used CRISPR-Cas9 technology to reintroduce the candidate qv18 molecular lesion in the sax-7(qv30) null mutant background ^27^. The resulting allele of mig-6, qv33, profoundly suppresses the sax-7(qv30) defects, reducing the percentage of affected animals from 90% to 30% (Fig. 1 A,B), indicating that the suppressor mutation is indeed an allele of the gene mig-6. Except for gonad defects (similarly found in previously reported mig-6 mutations) ^40^, single mutant mig-6(qv33) animals are fully viable and fertile, displaying a wild-type phenotype including for neurons ASH and ASI, overall nervous system morphology, body wall musculature, and pharynx (Figs. 1A,B, S2, S6). In contrast, null alleles of mig-6 are sterile and embryonic and larval lethal ^40^. Knockdown of mig-6 by RNA interference (RNAi) mimicked the effect of mig-6(qv33), significantly suppressing sax-7 neuronal defects (Fig. 1C). This result further confirms our molecular identification of the suppressor, and indicates that qv33 is a loss-of-function mutation.
The short isoform MIG-6S is key in neuronal maintenance, acting through its papilin cassette and lagrin repeats
The gene mig-6 encodes a short isoform, mig-6S, and a long isoform, mig-6L (Fig. 1D) ^40^. We used five other mig-6 alleles previously studied in the context of distal tip cell migration ^40^ to analyze their effect on *sax-7-*mediated neuronal maintenance (Fig. 1D’, F). An allele that specifically affects mig-6L, e1931, failed to suppress the neuronal maintenance defects in sax-7(qv30); mig-6(e1931) (Fig. 1F), indicating that mig-6L is not implicated in this context. In contrast, other mig-6 alleles that like qv33 affect both the short and the long isoforms (k177, ev700 and ev701) profoundly suppressed the ASH/ASI neuronal maintenance defects in sax-7(qv30); mig-6 double mutant animals (Fig. 1F). We further validated that mig-6S is the functional isoform in neuronal maintenance by performing rescue assays using a mig-6S transgene expressed under its endogenous promoter (minigene pZH125 ^40^). Since the loss of mig-6 suppresses sax-7 neuronal maintenance defects, restoration of mig-6 function in double mutant animals sax-7; mig-6 is expected to result in the reappearance of sax-7 defects. Transgenic sax-7(qv30); mig-6(qv33) animals carrying wild-type transgenic copies of mig-6S(+) showed partial but significant rescue (Fig. 1B). The semi-dominant behavior of mig-6 mutations described in other contexts ^40^ and in our analyses (below, Figs. 3,5) could explain the partial rescue. Collectively, these results firmly establish that mig-6S is central in neuronal maintenance.
mig-6 encodes the conserved extracellular matrix MIG-6/papilin, which is orthologous to Drosophila and vertebrate papilin ^36,40–43^. Papilin is important for proper organogenesis in flies ^36^ and for gonad and pharynx development in worms ^21,40,44^ but its role in the nervous system remains largely unknown. Recently papilin was isolated in a screen for brain morphogenesis mutants, but its function awaits study ^45^, and in C. elegans, loss of mig-6 was shown to affect PVD neuron 1o dendrite development ^46^. Little is known about the molecular mechanism of papilin function. MIG-6/papilin is a multidomain glycoprotein harboring thrombospondin type 1 (TSP1) repeats, cysteine rich lagrin repeats, and Kunitz protease inhibitor domains, among other domains (Fig. 1D’ ^36,43^, and belongs to the ADAMTSL family of proteins (A Disintegrin and Metalloproteinase with Thrombospondin motifs-Like). Papilins, as other ADAMTSL proteins, are structurally related to ADAMTS metalloproteinases but lack the catalytic domain characteristic of ADAMTS, and no catalytic activity has been reported ^47,48^. Importantly, ADAMTSL proteins are characterized by the “papilin cassette" (Fig. 1D’), a region containing TSP1 domains and an ADAMTS spacer (homologous to non-catalytic domains of ADAMTS metalloproteinases) ^36,43^. Yet, the papilin cassette present in Drosophila papilin has been shown, in vitro, to bind and inhibit the activity of a procollagen N-proteinase, a vertebrate ADAMTS ^36^. In the C. elegans gonad, mig-6 function influences the localization and levels of ADAMTS proteins MIG-17 and GON-1 ^21,40^. This raises the possibility that MIG-6/papilin may play important roles in the extracellular matrix via its papilin cassette.
As a first step to identify the critical domains of MIG-6S in neuronal maintenance, we modeled the molecular consequences of these tested mig-6 alleles on MIG-6S. The mig-6(qv33) mutation results in a substitution of a glycine to a glutamic acid at amino acid 664 (G664E), located in a TSP1 repeat in the papilin cassette. A sequence alignment as well as the predicted structure of MIG-6S, modeled using ColabFold (Fig. 1D”, S3), shows that G664 is a highly conserved residue located at the entrance of a beta-strand (Fig. 1D”’). Replacement of a glycine with a larger residue in the qv33 mutant is likely to cause a steric clash with the disulfide bond formed by residues C657 and C696 (Fig. 1D’”) and could alter a potential binding of MIG-6S to the ECM, or other interactions.
Similarly, other mig-6 alleles that suppress sax-7 defects (Fig. 1F) affect residues located in the papilin cassette or in the adjacent lagrin repeats (Fig. 1D’, D”). Indeed, mig-6(k177) (Y650D) missense allele impacts a residue in a TSP1 domain of the papilin cassette. In the ColabFold predicted structure of MIG-6S, the aromatic portion of Y650 interacts with both L673 and T648 (Fig. 1D’”). Consistently, homologs of MIG-6S typically possess either a tyrosine or a phenylalanine at position Y650 (Fig. 1D’”). Missense allele mig-6(ev701) (C848) affects a residue in a lagrin repeat of MIG-6S, where it forms a disulfide bridge with C833 (Fig. 1D’”), perhaps explaining the strong conservation of a cysteine residue at this position. Finally, missense allele ev700 (harboring substitutions G878E and C879Y) affects two residues located within the lagrin repeats cysteine-rich region of MIG-6S that are also well conserved, with C879 forming a disulfide bridge with C864 (Fig. 1D’”). Thus, mutation at G878 and C879 could perturb the pattern of disulfide bridges within this region. Finally, missense allele mig-6(sa580) (G965E) affects a glycine residue located in a lagrin domain, which could disrupt the adjacent cysteine residues involved in interactions with C949 and C973 (Fig. 1D’”). However, mig-6(sa580) does not supress sax-7 neuronal defects (Fig. 1F), highlighting the specific effects of distinct mig-6 mutations. In sum, these analyses support the idea that the papilin cassette and neighboring lagrin repeats are crucial for the function of MIG-6S in neuronal maintenance.
To experimentally validate which are the most critical regions of the MIG-6S protein in the context of neuronal maintenance, we carried out rescue assays using recombinant versions (Fig. 1E, S4). A recombinant mig-6S transgene lacking the sequence that encodes the C-terminal region of the protein, which contains the Kunitz domains, retained rescuing activity in sax-7(qv30); mig-6(qv33) double mutant animals with reappearance of neuronal disorganization, similar to the full length mig-6S(+) (compare to Fig. 1B). This supports that the Kunitz domains are not essential for MIG-6S function in neuronal maintenance. In contrast, a recombinant mig-6S transgene lacking the sequence that encodes the N-terminal region containing the papilin cassette and lagrin repeats failed to rescue the function of mig-6 in sax-7(qv30); mig-6(qv33) double mutants. These results, together with our analyses of a series of mutant alleles (molecular impact in Fig. 1D and functional consequences in Fig. 1F), demonstrate that the papilin cassette and the nearby lagrin repeats are the key domains for MIG-6S functionality in neuronal maintenance.
mig-6/papilin modulates neuronal maintenance post-embryonically and in specific neuronal contexts
Since several mig-6 mutations suppress the progressive head ganglia disorganization of sax-7 mutants, we asked whether losing mig-6 function after the development of ASH/ASI neurons would be sufficient to suppress sax-7 neuronal maintenance defects. As ASH/ASI neurons complete their development in embryogenesis, we depleted mig-6 function post-embryonically by feeding sax-7(qv30) animals with mig-6(RNAi) bacteria starting from the mid/late-L1 larval stage onward. Our results showed that the post-embryonic depletion of mig-6 function strongly suppressed sax-7 ASH/ASI neuronal maintenance defects (Fig. 1C), highlighting its post-developmental in maintaining ASH/ASI neuronal architecture.
We next explored if mig-6’s impact on neuronal maintenance is context-specific or generalized across the nervous system. sax-7 mutants are known to exhibit defects in maintaining axon positioning along the ventral nerve cord: axons of bilateral neurons PVQL/R initially develop normally, projecting ipsilaterally during embryogenesis, but later get displaced to the opposite fascicle of the ventral nerve cord in sax-7 animals ^25^, coinciding with remodeling of the underlying tissue during late first larval stage ^30,49^. We found that mig-6(qv33) partially suppresses these sax-7 axon “flip-over” defects (Fig. 2A, B), indicating that mig-6 participates in the maintenance of nerve cord organization as well.
The small secreted two-immunoglobulin proteins ZIG-3 and ZIG-4 also function to maintain axon position in the C. elegans ventral nerve cord ^30,31,33^, as does the giant basement membrane protein DIG-1, which is also required for ganglia maintenance ^28,29^. To determine if disrupted mig-6 function could also suppress the defective maintenance of axon position in these other known neuronal maintenance mutants, we generated mutant combinations between mig-6 and either zig-3 and zig-4, or dig-1. We found that mig-6(qv33) did not supress the axon flip-over defects in the double mutant of small secreted two-immunoglobulin proteins zig-3(tm924) zig-4(gk34) (Fig. 2C), highlighting the specificity of mig-6 effects. However, mig-6(qv33) did suppress axon flip-over in dig-1(ky188) mutants (ky188 is the dig-1 allele with the most penetrant axonal defects; Fig. 2D). As dig-1 mutants also exhibit defects in the maintenance of both tail and head neuronal ganglia organization, we examined the impact of mig-6(qv33) on dig-1 ganglia organization maintenance. We found that loss of mig-6 did not supress defective soma positioning of PVQ neurons in the tail ganglia of dig-1(ky188) mutants (ky188 display progressive and penetrant PVQ soma displacement; Fig. 2E), but did partially suppress head ganglia organization in dig-1(n1321) mutants (n1321 is the most severe dig-1 allele for head ganglia; Fig. 2F). These results indicate that the role of mig-6 in neuronal maintenance is specific and context-dependent, varying with the type of neuronal maintenance molecule affected and neuronal structure.
mig-6/papilin functions non-autonomously, together with mig-17/ADAMTS to impact neuronal maintenance
Given that disrupting the function of the extracellular matrix protein MIG-6/papilin counteracts the progressive neuronal disorganization observed in head ganglia and the nerve cord of sax-7 and dig-1 mutants, we hypothesized that the absence of functional MIG-6/papilin protein would affect the ECM surrounding these neuronal structures. In the developing gonad, mig-6 genetically interacts with mig-17/ADAMTS, which encodes a secreted metalloprotease of the ADAMTS family ^40^, thought to remodel the gonadal basement membrane ^21,50–53^. We therefore sought to investigate the functional relationship between mig-6/papilin and mig-17 in neuronal maintenance. We first examined mig-17(k174) putative null mutant animals ^54^ (null allele used throughout this study) and found that ASH and ASI neurons are normally positioned with respect to the nerve ring (Fig. 3A,C). mig-17 mutants exhibit an elongated pharynx ^55^ (Fig. S5), but this did not affect neuronal position with respect to body length. Indeed our data show that there is no difference in the relative positions of the nerve ring, or of ASH and ASI soma, between mutants with normal pharynx length (such as mig-6(qv33)) and mig-17 mutants (Fig. S5), indicating that neuronal positioning is independent from pharynx length. To test if loss of the ECM remodeling molecule mig-17/ADAMTS would impact the sax-7 head ganglia disorganization, we looked at double mutants lacking both sax-7 and mig-17. In double mutants sax-7; mig-17, only 27% of animals display head ganglia disorganization, compared to ~90% in sax-7 single mutants (Fig. 3A,C). This result shows that, similar to loss of mig-6, loss of mig-17 significantly supresses the neuronal maintenance defects in sax-7 mutants. In contrast, loss of another secreted ADAMTS metalloprotease, ADT-2/ADAMTS, involved in C. elegans body size regulation and cuticle structure ^56^, did not supress sax-7 defects nor enhance their suppression by mig-6 mutation (Fig. 3C). This highlights the specific role of mig-17/ADAMTS in neuronal maintenance.
To then determine whether mig-6 and mig-17 function in the same genetic pathway in this context, we constructed a triple sax-7; mig-6(qv33) mig-17(k174) mutant strain. We found that the simultaneous loss of mig-6 and mig-17 did not further enhance the suppression of neuronal maintenance defects in sax-7 mutants (Fig. 3C), suggesting that mig-6 and mig-17 may function in the same pathway to maintain neuronal architecture. To further probe the notion that mig-6 and mig-17 function collaboratively to impact neuronal maintenance, we analyzed the effect of partially losing the function of both genes using double heterozygous animals mig-6(qv33) mig-17(k174) / mig-6(+) mig-17(+), abbreviated as mig-6 mig-17/++. Single heterozygous animals mig-17(k174)/+ only slightly supressed sax-7 neuronal defects (Fig. 3C). Meanwhile, single heterozygous mig-6(qv33)/+ mildly suppressed sax-7 defects; Fig. 3C), consistent with observations that mig-6 alleles (ev700, ev701 and k177) behave in a semi-dominantly due to haploinsufficiency during gonad and PVD neuron development ^40,46^. Notably, in double heterozygous animals sax-7; mig-6 mig-17/++, the suppression of sax-7 defects was significantly greater, with only 69% of animals showing defects, compared to 81% in sax-7; mig-6/+ animals (Fig. 3C). This result indicates that the two extracellular matrix genes mig-6/papilin and mig-17/ADAMTS act within the same pathway to influence the long-term maintenance of neuronal organization.
Given that the neurons’ environment controls their maintenance, mig-6/papilin may be expected not to function from the neurons themselves. Indeed, extracellular matrix components, including MIG-6, are produced by mesodermal cells, particularly body wall muscles, and the epidermis ^21,40,44,57,58^. We thus generated transgenic sax-7; mig-6 double mutants animals expressing wild-type mig-6S under different tissue-specific promoters: Prgef-1 for neurons, Pdpy-7 for epidermis (hyp7), and Pmyo-3 for mesodermal cells (body wall muscles). Since the loss of mig-6 suppresses sax-7 neuronal maintenance defects, restoring mig-6 function is expected to lead to the reappearance of neuronal disorganization in sax-7; mig-6 double mutants. We found that expression of mig-6S(+) in the neurons or the epidermis did not rescue mig-6 function. In contrast, expression of mig-6S(+) in the body wall muscles in sax-7; mig-6 double mutant animals robustly rescued the neuronal maintenance defects, with an increase of neuronal defects from 29% up to 72%, depending on the transgenic line (Fig. 3D). This indicates that mig-6 functions cell non-autonomously from muscles to impact neuronal maintenance. As a control, we expressed these transgenes in wild-type animals and observed no neuronal defects (Fig. 3E), ruling out the possibility that mig-6S(+) overexpression by muscles induces artefactual neuronal disorganization. Overall, this confirms that the neuronal defects observed in transgenic animals expressing mig-6S(+) in body wall muscles represent bona fide rescue of mig-6 function.
We then investigated whether mig-17 is necessary for mig-6’s function in neuronal maintenance. To test whether the loss of mig-17 would affect the ability of mig-6S(+) transgene to rescue, we generated sax-7; mig-6 mig-17 triple mutants carrying transgenic lines expressing wild-type mig-6S(+) from body wall muscles (lines #17 and 18 used above, Fig. 3D). Unlike the successful rescue of mig-6 function observed in sax-7; mig-6 transgenic animals, the loss of mig-17 prevented the mig-6S(+) transgene from rescuing neuronal maintenance defects, as the transgenic triple mutant sax-7; mig-6 mig-17 animals did not exhibit a reappearance of neuronal defects (Fig. 3D). This may indicate that the normal function of MIG-6/papilin depends on MIG-17/ADAMTS, or alternatively, an excess of MIG-6S cannot compensate for the loss of MIG-17. In conclusion, we find that mig-6/papilin acts non-autonomously from muscles to suppress sax-7 neuronal maintenance defects in a manner that requires the function of mig-17/ADAMTS.
Loss of mig-6S/papilin results in increased EMB-9/collagen IV levels that accumulates as extracellular fibrotic-like structures
Given that MIG-6/papilin is an extracellular ADAMTS-like protein which interacts functionally with MIG-17/ADAMTS, we hypothesized that the mig-6 mutation may suppress sax-7 neuronal maintenance defects by modulating the extracellular environment, including nearby neurons. To directly test whether loss of mig-6 leads to changes in the extracellular matrix, we analyzed the distribution of a key ECM component, EMB-9/collagen IV α1 (hereafter referred to as ‘EMB-9/collagen IV’), known to genetically interact with mig-6 during gonadal development ^40^ or to be altered in the gonadal basement membrane in mig-6(RNAi)-treated animals ^21^. In C. elegans, collagen IV, similar to its vertebrate counterparts, is a heterotrimeric molecule consisting of two EMB-9 (α1-like) chains and one LET-2 (α2-like) chain, previously shown to colocalize ^59–62^. We therefore used a Pemb-9::EMB-9::mCherry fluorescent reporter ^63^ (gift from David Sherwood) to examine the distribution of collagen IV, focusing particularly on the head region (Fig. 4). In mig-6(qv33) mutants, like in the wild type, we observed collagen IV signal along the contour of the pharynx and the surface of body wall muscles (Fig. 4A, Fig. S6), corresponding to the basement membranes of these structures ^21^, as well as in spherical accumulations within muscle cells where collagen IV is produced. However, in mig-6 mutants, the overall abundance of EMB-9/collagen IV is higher compared to the wild type (Fig. 4A; see Fig. S7 for quantification of collagen IV in entire head region including muscle cells), including inside muscle cells. Moreover, mig-6 mutants display notable enrichments of collagen IV, often elongated, which we have termed “fibrotic-like structures” (Fig. 4A,B; Fig. S6). These fibrotic-like structures are extremely rare in the wild type and seen only in aging adults (5-day old adults), but are detected as early as the 1st larval stage in mig-6(qv33) mutants, and by the 4th larval stage and adulthood, 100% of the mig-6 mutants present fibrotic collagen IV (Fig. 4A,B). These fibrotic-like structures in mig-6 mutants are typically located in the posterior region of the head (Fig. 4C,D). Interestingly, our repeated observations of the same mig-6(qv33) animals in a longitudinal analysis over several days (Fig. 4E, n=8) show that the mig-6 mutants’ collagen IV fibrotic-like structures stably persist over time. Measuring the size of these structures at different developmental stages further confirmed that they lengthen in an age-progressive manner (Fig. 4F).
We next studied the localization of the collagen IV fibrotic-like structures that occur in mig-6 mutants. In C. elegans, collagen IV is produced by body wall muscles and other mesodermal cells, including the head mesodermal cell (hmc) and GLR glia ^64,65^. We thus generated a strain of mig-6(qv33) mutants carrying both the EMB-9::mCherry reporter and dnIs13 gly-18p::gfp ^66^ to simultaneously label collagen IV and anterior head wall muscles, the hmc, and GLR glia, respectively. Confocal microscopy revealed that collagen IV fibrotic-like structures in mig-6 mutants do not overlap with muscle cells, nor with hmc, nor the GLR glia, and are located extracellularly (Fig. 4I). In addition, spherical collagen IV deposits seen in body wall muscles, appear to be more intense in mig-6 mutants than in wild type (Fig. 4A; Fig. S6), and these are indeed intracellular (Fig. 4I).
To further support our findings, given that mCherry protein fusions can form aggregates ^67^, we employed an alternative multicopy reporter, EMB-9::Dendra2 ^63^ (gift of David Sherwood). With this reporter we also observed that EMB-9 accumulates as fibrotic-like structures in 33% of animals following mig-6(RNAi) knockdown (Fig. 4G). Importantly, using an endogenous CRISPR knock-in reporter for collagen IV ^21^, EMB-9::mNG, 90% of the mig-6(qv33) mutants show fibrotic-like structures (Fig. 4H).
Since collagen IV accumulates in mig-6 mutants, we assessed the stability of EMB-9::mCherry by fluorescence recovery after photobleaching (FRAP), which we performed on muscle and pharyngeal basement membranes that contain collagen IV, present in both the wild type and mig-6(qv33) mutants (Fig. S8A). Our FRAP measurements revealed no significant difference between mig-6 mutants and the wild type, with very limited collagen IV recovery across different time points (Fig. S8B), supporting the notion that loss of mig-6 does not affect collagen IV short-term dynamics per se, which is consistent with its described stable association with the gonadal basement membrane ^21^.
We further strengthened our findings that loss of mig-6 alters collagen IV levels and distribution by looking at collagen IV in other mig-6 loss-of-function backgrounds. We generated animals with the k177 allele of mig-6 (Y650D mutation in the same domain as qv33 G664E) carrying EMB-9::mCherry. Similar to qv33 animals, k177 mutants exhibit a significant accumulation of EMB-9/collagen IV, with approximately 80% of adult animals displaying fibrotic-like structures (Fig. 6A,B; Fig. S6). Similarly, RNAi-mediated knockdown of mig-6 resulted in 72% of adult animals exhibiting EMB-9::mCherry fibrotic-like structures (Fig. 4G). In contrast, specifical loss of mig-6L, using allele e1931, did not alter the collagen IV pattern nor led to fibrotic-like structures (Fig. 6A,B; Fig. S6). These results suggest that mig-6S, but not mig-6L, is essential for proper extracellular collagen IV organization in the head region. Overall, consistent with the findings of papilin affecting collagen IV in the gonadal basement membrane ^21^, we found that the formation of extracellular collagen IV fibrotic-like structures in the head region is a robust phenotype linked to the loss of mig-6S/papilin function.
mig-6/papilin affects the biomechanical properties of the animal’s tissues
Our findings demonstrate that disruption of MIG-6/papilin results in a dramatic collagen IV fibrotic phenotype (Fig. 4) and that the progressive neuronal disorganization in head ganglia and the nerve cord of sax-7 and dig-1 mutants is counteracted in mig-6 mutants (Fig. 1 and 2). We therefore hypothesized that the environment surrounding these neuronal structures may be modified in mig-6(qv33) mutants in such a way that results in enhanced maintenance of neuronal architecture. To start addressing this possibility, we characterized the biomechanical state of tissues in mig-6(qv33) mutants by measuring their viscoelasticity properties using Brillouin microscopy. This label-free imaging technique allows for the assessment of the viscoelastic properties of biological samples through photon–phonon scattering interactions ^68^. The key parameters measured are the Brillouin scattering induced frequency shift and linewidth, which provide information on the high-frequency longitudinal modulus and therefore the elastic and viscous properties of the sample, respectively ^69–71^. We first ensured that the refractive indexes of the head region were similar between mig-6 and wild-type animals at the examined ages (Fig. S9A), which is an important prerequisite to render the Brillouin microscopy results comparable to one another. Next, using a confocal Brillouin microscope ^72^, we imaged a head region at the level of the studied neuronal cell bodies, which includes other cells such as other neurons, muscles, epidermis, glia, as well as the associated basement membranes/ECM (Fig. 5A,B). Our findings show a significant decrease in Brillouin shift in mig-6 mutants compared to the wild type at the L4 larval stage (Fig. 5C,D; Fig. S9B), indicating a reduction in tissue elasticity. This change was not detected at the earlier L2 stage, suggesting that the loss of mig-6 affects tissue elasticity more prominently at later developmental stages. The decreased elasticity was most pronounced in the posterior zone of the region of interest (ROI), which exhibited lower Brillouin elastic contrast (Fig. S9C). Furthermore, Brillouin linewidth measurements revealed that tissue viscosity was also reduced in mig-6 mutants at both L2 and L4 stages (Fig. 5C,D; Fig. S9B). To assess tissue viscoelasticity in wild-type and mig-6 mutants, we measured the Brillouin loss tangent parameter ^73^, which showed decreased tissue viscoelasticity in mig-6 mutants compared to controls at both the L2 and L4 stages (Fig. 5C,D; Fig. S9B). These results demonstrate that MIG-6/papilin is essential for maintaining proper tissue mechanical properties in vivo during age-progression. Furthermore, as the altered biomechanical properties of the tissues in the head of mig-6 mutants are detected earlier than the appearance of ASH/ASI neuronal disorganization in sax-7 mutants, this suggests that properties of the underlying head ganglia environment play an important role in maintaining neuronal architecture over time.
mig-6/papilin and mig-17/ADAMTS function together to regulate extracellular collagen IV
Building on the previous finding that mig-6 genetically interacts with mig-17/ADAMTS to suppress sax-7 neuronal defects (Fig. 3), we investigated whether mig-6 and mig-17 also functionally interact in regulating collagen IV distribution. mig-17(k174) single mutants exhibit fibrotic-like structures in 75% of young adults and 95% of 2-day old adults (Fig. 6A,B; Fig. S6), similar to the phenotype observed in mig-6(qv33) and mig-6(k177) mutants. The simultaneous loss of mig-6 and mig-17 in double homozygous mig-6 mig-17 mutants did not enhance the collagen IV fibrotic-like structures compared to single mutants, neither in penetrance (Fig. 6B), nor in expressivity (Fig. 6D), consistent with the notion that the extracellular matrix genes mig-6 and mig-17 function within the same pathway to regulate collagen IV distribution.
To reinforce this conclusion, we assessed the effect of simultaneously losing a single functional copy of each gene, which can provide insights into genetic interactions (avoiding the ceiling effect, as heterozygous animals are much less penetrant for this phenotype). In mig-6(qv33)/+ single heterozygotes, we observed collagen IV fibrotic like-structures in approximately 10% of young adults, increasing to 75% of 2-day-old adults (Fig. 6A,B; Fig. S6), indicating a semi-dominant effect of mig-6 also for this phenotype. In mig-17(k174)/+ single heterozygotes, only around 10% of animals displayed fibrotic like-structures at both ages. Remarkably, in young adult double heterozygotes mig-6 mig-17/++, as many as 80% exhibited collagen IV fibrotic-like structures (increasing to 85% in 2-day-old adults; Fig. 6A,B; Fig. S6). Also, the number of fibrotic-like structures per animal strikingly increases in the double heterozygous mig-6 mig-17/++ animals compared to single heterozygous (Fig. 6C). Together, these results firmly establish that mig-6 and mig-17 functionally interact to regulate extracellular collagen IV.
MIG-17/ADAMTS has been hypothesized to function as a metalloprotease that degrades collagen IV ^74,75^. We thus asked whether overexpression of wild-type copies of mig-17(+) impacts the collagen IV phenotype of mig-6 mutants. For this we used functional transgene Pmig-17::mig-17(+)::gfp (Nishiwaki et al, 2000; Jafari et al, 2010) (Fig. 6A and Fig. S6), combining the integrated transgene (evIs213, gift of Joe Culotti) with mig-6(qv33). We found that while the percentage of mig-6 animals displaying fibrotic-like structures was unchanged (Fig. 6B), the number of fibrotic-like structures per young adult animal significantly decreases in mig-6 mutants overexpressing the mig-17(+) transgene (Fig. 6D). This result indicates that the collagen IV defects of mig-6 mutants can be partially offset by mig-17/ADAMTS overexpression.
As mig-6 mutants display a fibrotic collagen IV phenotype, and the overexpression of mig-17(+) partially suppresses this defect (Fig. 6D), we wondered whether levels of MIG-17/ADAMTS may be lowered in mig-6 mutants. To test this idea, we examined the distribution of MIG-17/ADAMTS in mig-6(qv33) mutants using P*mig-17::*MIG-17::GFP (evIs213). We found that instead of being decreased, MIG-17 is in fact upregulated in mig-6 mutants (Fig. 6E), with new enrichments in different head regions compared to wild type. Given that increased mig-17(+) levels can at least partially suppress mig-6 mutants (Fig. 6D), and that MIG-17 levels are elevated in mig-6 mutants (Fig. 6E), together these results suggest that MIG-17/ADAMTS’s function requires functional MIG-6/papilin to robustly ensure normal collagen IV distribution.
Suppression of sax-7 neuronal maintenance defects upon loss of mig-6/papilin and mig-17/ADAMTS depends on collagen IV levels and cross-linking
Because mig-6 and mig-17 cooperate to regulate extracellular collagen IV (Fig. 6A–D) and maintain neuronal organization in sax-7 mutants (Fig. 3A,B,C), we asked if the suppression of sax-7 neuronal maintenance defects upon loss of mig-6 or mig-17 is linked to changes in collagen IV patterning in the ECM. We assessed the state of collagen IV distribution in the sax-7 mutant background and found that sax-7 single mutants behave like the wild type in this regard (Fig. 6F,G; S6). Also, double mutants sax-7; mig-6(qv33) and sax-7; mig-6(k177), as well as sax-7; mig-17(k174) displayed a fibrotic-like structure phenotype like that of the respective mig-6 or mig-17 single mutants (Fig. 6F,G; S6). That sax-7; mig-6 and sax-7; mig-17 double mutants exhibit modified collagen IV pattern (Fig. 6F,G) and maintain organized head ganglia (Fig. 3C) is consistent with the notion that the state of the ECM plays a role in maintaining neuronal architecture. In line with this, mig-6L-specific allele e1931 does not affect the pattern of collagen IV (normal levels and no fibrotic structures; Fig. 6A,B; S6) and does not suppress the neuronal maintenance defects of sax-7 mutants (Fig. 1F).
We then investigated whether collagen IV levels and distribution contribute to the maintained neuronal organization of double mutants sax-7; mig-6 and sax-7; mig-17. We depleted emb-9/collagen IV by RNAi treatment of animals from the 1st larval stage and examined head ganglia organization in adults. This emb-9(RNAi) knockdown effectively depleted EMB-9/collagen IV levels (Fig. 7A, Fig. S10A), and importantly, did not affect neuronal organization in wild-type animals, nor in single mutants. In contrast, depleting collagen IV reversed the suppression of sax-7 neuronal defects by mig-6 or mig-17 mutation. Indeed, double mutant animals sax-7; mig-6 and sax-7; mig-17 showed increased neuronal disorganization upon emb-9(RNAi) (Fig. 7B). This result indicates that collagen IV levels are key for the suppression of sax-7 neuronal defects by the loss of mig-6 or mig-17. We then tested whether a higher level of collagen IV could mimic the effect of mig-6 loss of function in suppressing sax-7 neuronal maintenance defects. We found that sax-7 animals overexpressing transgene emb-9(+) (in sax-7; qyIs46 animals carrying the multicopy transgene P*emb-9::*EMB-9::mCherry) did not show suppression of neuronal maintenance defects (Fig. 7C). Thus, while sustained levels of collagen IV are required to suppress sax-7 neuronal defects, elevated collagen IV level per se is insufficient to ensure neuronal maintenance.
We therefore investigated whether collagen IV organization plays a role in neuronal maintenance as well. Collagen IV molecules form complex crosslinked networks involving dimerization through their NC1 domain ^65,76,77^. The extracellular enzyme peroxidasin catalyzes sulfilimine S=N bonds between collagen IV NC1 domains ^76,78^, which are essential for collagen IV networks and basement membrane integrity ^79–82^. The C. elegans genome encodes two peroxidasins, of which PXN-2/peroxidasin is known for its effects on the ECM and genetic interactions with collagen IV genes ^83^. We examined the pattern of PXN-2/peroxidasin using a knock-in fluorescent reporter mNeonGreen::PXN-2 (driven under the pxn-2 promoter ^21^, and found that the expression of PXN-2/peroxidasin is upregulated in mig-6(qv33) mutants compared to wild type (Fig. 7D), suggesting that mig-6 is implicated in the regulation of peroxidasin 2 levels in the ECM. Knockdown of pxn-2/peroxidasin by RNAi (from the 1st larval stage) did not significantly lower the penetrance of fibrotic-like structures (Fig. S10B), but significantly reduced the number of fibrotic-like structures (Fig. 7E,F), and led to a striking increase of fragmented fibrotic collagen IV (Fig. 7E,G). Importantly, pxn-2(RNAi) knockdown reverses the suppression of sax-7 mutants’ neuronal defects by loss of mig-6 or mig-17 (Fig. 7H). Together, these results highlight that the function of MIG-6/papilin and MIG-17/ADAMTS in neuronal maintenance is dependent on collagen IV and its crosslinking by the peroxidasin enzyme. Further, they support the notion that the elevated levels of crosslinked collagen IV in the ECM of mig-6 mutants contribute to stabilizing neuronal position and maintaining neuronal architecture in sax-7 mutants (Fig. 7I).
Loss of mig-6/papilin counteracts neuronal disorganization induced by increased mechanical stress
Since loss of mig-6 function positively impacts the maintenance of neuronal organization in animals lacking the cell adhesion molecule SAX-7/L1CAM, we hypothesized that it may also support neuronal architecture in otherwise wild-type animals that experience increased internal mechanical stress. We used the distinct locomotion patterns of C. elegans to probe this question. In liquid media, C. elegans swims, whereas on solid media, it crawls. Swimming and crawling differ in neuromuscular activity and speed, with swimming being faster ^84,85^. The associated exerted forces are also different: when the worm crawls on solid media, the forces exerted on the worm’s cuticle are higher than when swimming in liquid ^86–88^. By contrast, swimming worms perform many more body bends, with more numerous body wall muscles contractions, which is expected to result in higher mechanical stress on internally located neurons (e.g., in head ganglia) compared to the slower movements of worms crawling on solid media. We therefore subjected worms to continuous swimming in liquid culture from the time of hatching to adulthood, and assessed the position of sensory neurons ASH and ASI. Compared to the neurons in animals grown on solid medium, neuronal position in wild-type animals changed significantly when grown in liquid medium for 5 days, exhibiting a significant posterior displacement (Fig. 7J). However, mig-6(qv33) mutant animals raised in liquid medium showed a significant decrease in neuronal displacement (Fig. 7J), indicating that loss of mig-6/papilin function results in enhanced maintenance of neuronal organization upon high internal mechanical stress.
Because collagen IV is required for the stabilizing effect of sax-7 mutants neuronal organization by loss of mig-6 function, we examined the state of collagen IV in swimming animals. We observed that mig-6 mutants display an altered collagen IV pattern (similar to that of mig-6 mutants grown on solid medium), with the presence of fibrotic-like structures, increasing in penetrance from day 3 to day 5 post L1 hatching (Fig. 7K). We then asked if collagen IV was required for the neuronal protective effect conferred by the mig-6 mutation in otherwise wild-type animals when grown in liquid. Depleting collagen IV by emb-9(RNAi) of swimming animals significantly weakened the mig-6-mediated stabilizing effect of neuronal organization of ASH and ASI neurons (Fig. 7L). This result indicates that collagen IV is critical in the neuronal protective mechanism involving mig-6/papilin in conditions of increased mechanical stress.
DISCUSSION
Neuronal architecture established embryonically must persist throughout life to ensure nervous system function. However, the mechanisms sustaining neuronal organization over the long term remains poorly understood. This work uncovers a novel mechanism where ECM dynamics plays a critical role in maintaining neuronal architecture. Through a multidisciplinary approach, integrating forward genetic screening, incisive molecular genetic analysis, structural molecular predictions, quantitative live imaging, and measurement of biomechanical properties by Brillouin microscopy, we have identified the evolutionarily conserved extracellular matrix protein MIG-6/papilin as a key regulator of the long-term maintenance of the neuronal architecture. We show that MIG-6/papilin impacts neuronal maintenance by modulating the animal’s tissues biomechanical properties and remodeling the extracellular network of collagen IV, which is a major component of the basement membranes, including those surrounding neuronal assemblies. We also find that ECM metalloproteinase MIG-17/ADAMTS is important for sustaining neuronal architecture, and functionally cooperates with MIG-6/papilin in ECM remodeling in order to enable the long-term stability of neuronal architecture. Both the abundance and the cross-linking of collagen IV networks are essential for the MIG-6/papilin-remodeled ECM state that enables the maintenance of neuronal structures. Thus, this work reveals a previously unknown mechanism by which ECM remodeling enables the preservation of neuronal architecture (Fig. 8), in the face of age-progressive stresses, to preserve continuous neural function.
MIG-6/papilin is expressed throughout life, from embryogenesis to adulthood ^21,36,40,57,58^, in dynamic patterns that may reflect its requirements at the different life stages. Papilin indeed plays key developmental roles: the lack of papilin results in embryonic lethality in null mutant worms and in RNAi-depleted flies ^36,40^. Papilin is also required for organogenesis in flies ^36^, distal tip cell migration of the developing C. elegans gonad ^40^, as well as for enlargement of the gonad and the pharynx during C. elegans’ growth ^21,44^. A role for papilin in the nervous system has remained largely unexplored. There is one study in C. elegans showing that papilin participates axon guidance of the neuron ALA, which impacts primary dendrite development of the neuron PVD ^46^. In Drosophila, a recent report on a screen for regulators of central nervous system morphology mentions a papilin mutant found to have a misshapen central nervous system ^45^, which awaits further analysis. In our study, we isolated the mutation mig-6(qv33) in a screen for animals that suppress the age-progressive disorganization of sax-7/L1CAM mutants. Similar to other mig-6 mutations, mig-6(qv33) mutant animals have gonad abnormalities, but otherwise display normal body morphology (i.e., normal musculature, pharynx, epidermis, and overall neuronal architecture). We uncovered a post-developmental role of mig-6 in maintaining the positioning of neurons ASH and ASI, since depletion of mig-6 function by RNAi treatment initiated from the first larval stage onwards suppressed the ASH and ASI neurons’ defects that progressively accumulate in sax-7 null mutants, after having normally developed during embryogenesis.
mig-6/papilin encodes two isoforms, and our allelic series analysis and rescue assays demonstrate that the short isoform, MIG-6S, is active in neuronal maintenance, while MIG-6L is dispensable in this context. Isoform-specific roles of mig-6 have been previously described in the gonad development ^40^ and ALA-PVD neuronal patterning ^46^. MIG-6S belongs to the ADAMTS-like ECM protein family, and consists of several domains, including the papilin cassette with its TSP1 repeats and the ADAMTS spacer, followed by numerous cysteine-rich lagrin repeats, and Kunitz domains. Our genetic and protein-domain analyses, combined with molecular predictions, point to the papilin cassette and the closest lagrin repeats as being critical for MIG-6S function in neuronal maintenance. Indeed, disruption of mig-6S function by mig-6(qv33) or other mutations affecting the papilin cassette/adjacent lagrin repeats suppressed the age-progressive neuronal disorganization of sax-7/L1CAM mutants, but mig-6(sa580) that affects a different lagrin repeat did not impact the neuronal maintenance. Allele-specific effects of mutations affecting this region of the protein have also been described in the context of pharynx growth, where mutants mig-6(sa580) do display a twisted pharynx, but the mutations mig-6(k177, ev700, or ev701), which like mig-6(qv33) affect residues in more N-terminally located TSP1 domains or lagrin repeats, do not affect pharynx development ^44^. These allele-specific defects likely reflects the complexity of interactions of this multidomain MIG-6/papilin. Interestingly, mig-6(qv33) is a semi-dominant allele in neuronal maintenance, indicating that a minimum level of MIG-6/Papilin is required for proper function in this context. The mig-6 locus was similarly described as haploinsufficient in gonad and ALA-PVD neuron development ^40,46^.
In both C. elegans and Drosophila, papilin is expressed by cells in charge of producing the ECM/basement membrane components, such as body wall muscles and epidermis in the worm, and hemocytes in flies. Indeed, we found that expression of MIG-6S from body wall muscles rescued its function in neuronal maintenance. Interestingly, MIG-6S/papilin itself localizes to the basement membrane of several organs, including the gonad, the pharynx, the intestine ^21,40^, as well as nerve structures (e.g., nerve tracts ^46^) in C. elegans larvae and adults. Similarly, in Drosophila, papilin localizes to the basement membranes, including those enveloping the central and peripheral nervous system of both larvae and adults ^36^. Interestingly, publicly available data shows that papilin is also expressed in central nervous system of adult mice (Allen Atlas, https://portal.brain-map.org/), suggesting papilin may function in the adult mammalian brain as well.
How might the basement membrane protein MIG-6/papilin regulate neuronal maintenance? The extent to which ECM remodeling determines the long-term preservation of the neuronal architecture laid out earlier in development is only beginning to be probed. We report that the role of the extracellular papilin MIG-6 and the ADAMTS protease MIG-17 in maintaining neuronal organization in C. elegans is through their cooperative function in regulating collagen IV. Papilin is a component of basement membranes but appears to have essential roles in their assembly or maintenance, since all of the mig-6 mutants analyzed and mig-6(RNAi) treated animals display continuous basement membranes surrounding the pharynx, body wall muscles and the gonad, similar to wild type (this study, ^21^). Rather, papilin appears to affect specifically the remodeling of basement membranes, as disruption of MIG-6/papilin results in a dramatic build-up of extracellular collagen IV, a major component of ECM/basement membranes, indicating that MIG-6/papilin regulates collagen IV removal and distribution in the ECM. In mig-6S mutants and mig-6(RNAi) depleted animals, collagen IV accumulation is visible during larval stages and increases with age (both in terms of penetrance and of the extent of collagen build-up per animal), including within given individual animals as shown by our longitudinal analysis. Moreover, we observed an increase in intracellular collagen IV levels in body wall muscles, which produce both ECM components and MIG-6/papilin, suggesting that MIG-6/papilin may also impact collagen IV synthesis or degradation. RNAi-depletion of mig-6 also results in collagen IV accumulation in the gonadal basement membrane ^21^.
MIG-6/papilin is an ADAMTS-like protein, sharing structural similarities with ADAMTS secreted ECM metalloproteinases, but lacking a catalytic domain. Its biochemical function is unclear, but Drosophila papilin can bind a procollagen N-proteinase ADAMTS in vitro, inhibiting its activity non-competitively, without directly interfering with the enzyme’s catalytic site ^36^. The papilin cassette alone could also inhibit the procollagen N-proteinase. The ‘papilin cassette’ in the papilin ADAMTS-like proteins is important for binding to ECM ^89^, and papilin domains often interact with ADAMTS proteases also containing a papilin cassette ^36^, further supporting a regulatory role of this key region in ECM remodeling. MIG-17 is an atypical ADAMTS enzyme as it lacks TSP1 domains and thus a papilin cassette; yet MIG-17 is classified as belonging to the ADAMTS family based on the other significant structural similarities to ADAMTS proteins ^54,90^. Our genetic and molecular analyses revealed that ADAMTS-like protein MIG-6/papilin and ADAMTS metalloproteinase MIG-17 function within the same pathway to regulate ECM remodelling. Indeed, (i) the loss of mig-17 mirrors the mig-6S loss-of-function phenotype of extracellular collagen IV build up; (ii) the simultaneous loss of both genes in double homozygous mutant animals does not enhance the collagen IV fibrotic phenotype, and (iii) loosing half of the function of both genes in double heterozygous animals strongly enhances the defects. Although they function together to promote removal of extracellular collagen IV, mig-17 and mig-6S mutants do have phenotypic differences, notably relating to pharynx development; also, mig-6S mutants display a higher level of intracellular collagen IV in muscle cells, while mig-17 mutants do not. As a note, another genetic lesion of mig-17, ola226, was reported to have extracellular collagen IV accumulation in the head region ^51^. In sum, our data support the notion that MIG-6/papilin and MIG-17/ADAMTS functionally cooperate to regulate collagen IV remodeling. MIG-17/ADAMTS has been hypothesized to possess proteolytic activity toward collagen IV, based on studies both in C. elegans and Drosophila ^74,75,91^. It is thus conceivable that the build-up of collagen IV in the ECM of mig-6 mutants could result from MIG-17 being inhibited or less efficient, especially since we found that MIG-17 levels are increased in the head region of mig-6 mutant animals. We directly tested this by overexpression of functional MIG-17/ADAMTS, which did not reverse the collagen IV fibrotic phenotype in mig-6 mutants, suggesting that MIG-17/ADAMTS is in its active form in mig-6S mutants yet unable to degrade collagen IV, perhaps due to its high degree of crosslinking ^92–94^. In this scenario, MIG-6 regulates the level and activity of an ADAMTS through its impact on the ECM state.
Interestingly, we show that MIG-6/papilin influences the levels and distribution of MIG-17/ADAMTS and of the extracellular collagen IV crosslinking enzyme PXN-2/peroxidasin in the vicinity of the affected neuronal structures. Other studies have also documented that MIG-6/papilin affects the distribution of MIG-17 in the basement membrane of the developing gonad ^40^, and mig-6 depletion by RNAi increased the levels of the ADAMTS proteinases MIG-17 and GON-1, and of PXN-2/peroxidasin-2 in the gonadal basement membrane ^21^. Whether this involves a physical interaction (direct or indirect) between MIG-6/papilin and these proteins is to be determined. Regardless, these observations together suggest that papilin might play a broad role in collagen IV/ECM remodeling. Importantly, we show that loss of mig-6S or loss of mig-17 profoundly suppresses the neuronal maintenance defects that occur in sax-7/L1CAM mutants. Furthermore, losing the function of both MIG-6 and MIG-17 in homozygous double mutant animals did not enhance the suppression of the neuronal maintenance defects of sax-7 mutants, and loss of one copy of each gene in double heterozygous animals significantly enhanced the suppression compared to each heterozygous single mutant. These observations are consistent with the notion that MIG-6 and MIG-17 function in the same pathway to impact the maintenance of neuronal architecture. Evidence for a functional relationship between MIG-6 and MIG-17 exists also in the context of the C. elegans developing gonad, where mig-6 and mig-17 genetically interact ^40^. Importantly, MIG-17 is not involved in ALA-PVD neurons patterning, indicating that MIG-6/Papilin operates through distinct mechanisms depending on the biological context, which is consistent with the specificity of defects displayed by different alleles affecting mig-6S, possibly interacting with distinct functional partners through distinct regions of this multidomain protein.
At the level of multicellular neuronal structures, such as ganglia or nerve cords, a delicate balance must exist between ECM stability, which preserves the architecture of the existing neuronal structures, and ECM remodeling, which accommodates growth of the neuronal structures during post-natal life, as well as adapting to shape changes that accompany the animal’s movements. The shared fibrotic collagen IV phenotype between mig-6S and mig-17 mutants suggests that the altered state of collagen IV in these two mutants contributes to their ability to sustain neuronal architecture in sax-7 mutants. An excess of crosslinked collagen IV may reinforce the integrity of the basement membrane, thereby supporting the maintenance of neuronal organization. We favor a model in which enhanced basement membrane integrity leads to maintained neuronal architecture for several reasons. First, the mig-6 and mig-17 mutations that do suppress sax-7-neuronal disorganization display a dramatic accumulation of extracellular collagen IV. Second, both collagen IV abundance and its crosslinking are required for neuronal maintenance, as reducing collagen IV by emb-9(RNAi) significantly reversed the stabilizing effect brought about loss of MIG-6S or of MIG-17, as does reducing the crosslinking of collagen IV by RNAi knockdown of PXN-2/peroxidasin ^95^. Collagen IV was also key in the role of MIG-6/papilin in modulating the response of neuronal architecture to heightened mechanical stress, as loss of mig-6 was protective of head ganglia organization in animals subjected to swimming which leads to increased mechanical stress on the nervous system, due to the constant and rapid swimming muscle contractions was also dependent on collagen IV levels. Collectively, these findings underscore that extracellular collagen IV networks are key in neuronal maintenance. The fibrotic-like structures displayed by mig-6 and mig-17 mutants are unlikely directly involved in stabilizing neuronal architecture; rather, these fibrotic accumulations are the most obvious manifestations of dysregulated ECM remodelling, which also affects the basement membranes surrounding neuronal structures in mig-6 and mig-17 mutants.
Collagen IV, thanks to its unique ability to form intermolecular covalent bonds, provides the basement membrane with the capacity to withstand mechanical stress ^76,96^. Thus, we characterized the mechanical properties that result from the loss of functional MIG-6/papilin, more specifically, by analyzing the high-frequency longitudinal modulus of tissues, using Brillouin microscopy ^71^. We imaged an area neighboring the neurons under study, and compared mechanical properties of mig-6 mutants and wild-type animals at two ages, earlier in larval life, and just before becoming adults. The area imaged, located in the posterior region of the animal’s head, comprises several cell types, including neurons, muscles, glia, pharynx, and their ECM. While the contribution of each individual adjacent cell and of the local ECMs to the measured mechanical proprieties cannot be reliably discriminated in intact animals with the current resolution of the Brillouin microscope, ECM including basement membranes, is known to exert a key influence on tissue biomechanics ^97–99^. Thus, tissue viscoelastic properties are significantly determined by the ECM ^97,99^. Our Brillouin spectral analysis revealed that loss of MIG-6/papilin results in altered biomechanical properties in the head region which houses the neuronal ganglia we primarily analyze. Collectively, the imaged tissues and associated ECMs in mig-6 mutants have reduced viscosity and elasticity, indicating impaired viscoelastic properties. Importantly, cellular viscoelasticity is a regulator of cell behavior, associated with both physiological and pathological states across species ^97,99^. Thus, having been able to capture changes that inform on the viscoelastic properties of animals lacking MIG-6/papilin is a key finding, especially since few such in vivo measurements have been achieved to date ^88,100^.
The viscosity of a substrate is known to influence cell migration, with cells from normal tissue and tumor cells both exhibiting increased migration speed on highly viscous substrates or extracellular fluids ^101 {Bera, 2022 #723 102^. Also, both higher and lower cellular elasticity are linked to the motility of cancer cells ^103–105^. Conversely, cell adhesion can occur on the surface of low viscosity liquids ^106,107^. Thus, the decreased viscosity of mig-6 mutants may somehow, possibly via distinct cell-ECM interactions, result in enhanced cell adhesion enabling neurons to maintain their normal architecture. In addition, mig-6 mutants present a decrease in loss tangent that translates into decreased viscoelasticity, suggesting that their tissues exhibit more solid-like properties with reduced energy dissipation ^73^. Tissues and matrix mechanics are sensed by cells and converted into chemical signals through mechanotransduction ^97,108^. Thus, the decreased viscoelasticty in mig-6 mutants, and the proposed associated reduction in energy dissipation, could modulate mechanosensing and regulate cellular responses ^109,110^, to better preserve tissue shape and maintain neuronal architecture. Indeed, this may be related to the altered collagen IV levels, organization, and remodeling that we uncovered in mig-6 mutants, which could profoundly impact the overall ECM composition and organization. A parallel could be drawn with the excessive production of ECM components in tissue fibrosis, which results in decreased viscoelasticity ^108^. Such fibrotic states also lead to progressive matrix stiffening ^108^. The build-up of collagen IV occurring in mig-6 mutants, and that depleting the crosslinking enzyme peroxidasin/PXN-2 attenuated their fibrotic state, suggests that collagen IV molecules in mig-6 mutants have increased covalent sulfilimine cross-links, which could lead to increased ECM stiffness ^81,111^, and consequently, a likely decreased flexibility. Given the expected increased stiffness in mig-6 mutants and the similar altered remodeling of ECM collagen IV in double sax-7; mig-6 mutant animals, the mechanical proprieties arising from loss of mig-6 could help maintain neuronal architecture through increased stiffness.
Overall, we propose that the animal’s biomechanical changes resulting from the loss of MIG-6/papilin are linked to their altered ECM state. The differences in biomechanical properties are likely to bring about changes in ECM-neuron interactions, and/or in the state of neurons, such that neuronal architecture is preserved, even in the absence of SAX-7/L1CAM, or in conditions of heightened physical stress from the incessant muscle contractions of continuous swimming. Future studies could further elucidate the underpinnings of this remarkable state resulting from changes in ECM remodeling by the conserved ECM regulator MIG-6/papilin, which safeguards neuronal architecture during post-natal life and into adulthood.
Interestingly, while MIG-6/papilin plays a crucial role in defining the state of the ECM, its effects are specific to the precise molecular landscapes in distinct neuronal structures of the animal. For instance, we found that whereas loss of mig-6 suppresses the maintenance defects of axon position in the ventral nerve cord that are caused by the loss of adhesion molecule SAX-7/L1CAM, or by loss of basement membrane protein DIG-1, it fails to suppress similar maintenance defects of the same axons when caused by the loss of two-Ig domain proteins ZIG-3 and ZIG-4. Similarly, whereas loss of mig-6 suppresses the head ganglia defects in both sax-7/L1CAM and dig-1 mutants, it had no effect on tail ganglia maintenance defects displayed by dig-1 mutants. These observations underscore the complexity of the molecular interactions involving distinct ECM networks that surround different neuronal assemblies. The specificity of MIG-6/papilin’s action is also evident in the different developmental consequences of mig-6 mutations across different contexts, including the gonad, the pharynx, and the ALA-PVD neurons in the lateral nerve tract. This specificity is also reflected in its functional interaction with MIG-17/ADAMTS, which affects head ganglia maintenance and distal tip cell migration (this work,^40^), but not for ALA-PVD neuronal patterning ^46^. Future studies will help elucidate the interactions among other ECM components that may participate in the remodeling process orchestrated by papilin.
The extracellular matrix (ECM) has emerged as a key regulator of nervous system development and maintenance across diverse species ^112–114^. In C. elegans, the ECM modulates synaptic development ^115^, as well as synaptic maintenance, with collagen IV and metalloproteinase GON-1 being implicated in sustaining synapse morphology of neuromuscular junctions ^116 117^. In the mammalian central nervous system, the ECM is a large part of the neural tissue and serves various functions ranging from supporting cell migration, to regulating synaptic transmission and plasticity, to actively modulating the neural tissue after injury. In particular, the perineuronal nets (PNNs), a specialized form of ECM surrounding dendritic spines, have been shown to be dynamically regulated, impacting both structural and functional plasticity ^118^. The ECM composition of PNNs is regulated by the expression of proteases that target distinct PNN, enabling the transition from states of plasticity to stability ^119^. Disruptions in PNN composition is linked to neurodegenerative diseases ^13,14^. Collagen IV is a well-conserved component of basement membranes, including in the vertebrate central nervous system. It is conceivable that functional interactions between papilin, ADAMTS metalloproteinases and ECM molecules, similar to those described in this study, may also occur in mammals to maintain neuronal architecture throughout life.
Neuronal structures need to withstand deformations caused by the animal’s growth and body movements to prevent structural damage to neural circuits. How multicellular neuronal assemblies endure mechanical stress to sustain their architecture on the long term remains poorly understood. This work provides a mechanism by which the regulation of ECM remodeling enables and supports the maintenance of neuronal architecture postnatally and into adulthood. Other mechanisms previously described to critically impact the maintenance of neuronal architecture also rely on non-cell-autonomous biological functions. For instance, the secreted immunoglobulin proteins ZIG-3 and ZIG-4 are thought to stabilize axons positioning by modulating inter-axon adhesive properties ^30,31^. The cell adhesion molecule SAX-7/L1CAM mediates cell surface homophilic and heterophilic interactions between neurons and its neighboring cells (e.g, other neurons or epidermal) ^25,27,46^. The secreted basement membrane protein DIG-1 is proposed to bridge interactions between the basement membranes ensheathing neuronal structures and adjoining muscle cells ^28^. Collagen IV and ADAMTS/GON-1 ensures the maintenance of synaptic morphology at the neuromuscular junction ^116,117^. MIG-17/ADAMTS maintains synapse location and morphology during post-embryonic growth by modulating muscle basement membrane, which impacts interactions between epidermis, glia, and the associated synapses ^51,120^. The two-immunoglobulin domain protein ZIG-10 expressed on the epidermis underlying the nerve cord maintains synaptic density as the animal grows ^121^. More recently, the interplay between epithelial cells, their ECM, cell junctions and glial cells was shown to ensure the preservation of glia morphology in the face of environmental challenges, which in turn protects the associated neuron’s shape and function ^88,122^. Finally, cytoskeletal components too can act non-cell-autonomously from the underlying epidermis embedding the axon of a neuron to preserve its integrity ^123.^. Thus, the combined actions of both intrinsic and extrinsic mechanisms safeguard the intricate multicellular structures of the nervous system. Understanding general principles governing the long-term maintenance of the neuronal architectures underlying neural circuits is crucial for elucidating the bases of neurodegenerative conditions.
Supplementary Material
Supplement 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1van Dyck L. I. & Morrow E. M. Genetic control of postnatal human brain growth. Current opinion in neurology 30, 114–124 (2017).27898583 10.1097/WCO.0000000000000405 PMC 5340196 · doi ↗ · pubmed ↗
- 2Heckman E. L. & Doe C. Q. Establishment and maintenance of neural circuit architecture. Journal of Neuroscience 41, 1119–1129 (2021).33568445 10.1523/JNEUROSCI.1143-20.2020 PMC 7888231 · doi ↗ · pubmed ↗
- 3Bénard C. & Hobert O. Looking beyond development: maintaining nervous system architecture. Current topics in developmental biology 87, 175–194 (2009).19427520 10.1016/S 0070-2153(09)01206-X · doi ↗ · pubmed ↗
- 4Sultana O. F., Bandaru M., Islam M. A. & Reddy P. H. Unraveling the complexity of human brain: Structure, function in healthy and disease states. Ageing Research Reviews 100, 102414 (2024).39002647 10.1016/j.arr.2024.102414 PMC 11384519 · doi ↗ · pubmed ↗
- 5Hemphill M. A., Dauth S., Yu C. J., Dabiri B. E. & Parker K. K. Traumatic brain injury and the neuronal microenvironment: a potential role for neuropathological mechanotransduction. Neuron 85, 1177–1192 (2015).25789754 10.1016/j.neuron.2015.02.041 · doi ↗ · pubmed ↗
- 6Lin Y.-C. & Koleske A. J. Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annual review of neuroscience 33, 349–378 (2010).10.1146/annurev-neuro-060909-153204 PMC 306338920367247 · doi ↗ · pubmed ↗
- 7Mariano V., Domínguez-Iturza N., Neukomm L. J. & Bagni C. Maintenance mechanisms of circuit-integrated axons. Current opinion in neurobiology 53, 162–173 (2018).30241058 10.1016/j.conb.2018.08.007 · doi ↗ · pubmed ↗
- 8Sauerbeck A. D. SEQUIN multiscale imaging of mammalian central synapses reveals loss of synaptic connectivity resulting from diffuse traumatic brain injury. Neuron 107, 257–273. e 255 (2020).32392471 10.1016/j.neuron.2020.04.012PMC 7381374 · doi ↗ · pubmed ↗