Atopic skin inflammation promotes systemic anaphylactic responses via IL-13 signaling in conventional dendritic cells
Yasuyo Harada, Takanori Sasaki, Kazushige Obata-Ninomiya, Takahiro Matsuyama, Satoshi Ueha, Shigeyuki Shichino, Takashi Watanabe, Wu Bin, Shuhei Ogawa, Sewon Ki, Yoshie Suzuki, Hideki Ueno, Steven F. Ziegler, Hiromasa Inoue, Peter D. Burrows, Kenneth Murphy, Brian S. Kim

TL;DR
Skin inflammation in atopic dermatitis can lead to severe allergic reactions through IL-13 signaling in specific immune cells.
Contribution
The study identifies dendritic cells as key mediators of IgE-mediated anaphylaxis via IL-13 signaling.
Findings
Dendritic cells, not T or B cells, are critical for high-affinity IgE production in anaphylactic responses.
IL-13 signaling in cDC2s enhances MHC class II and CD301b expression, promoting type 2 immune responses.
Similar IL-13R-expressing dendritic cells were found in patients with allergic conditions and a history of AD.
Abstract
Cutaneous allergen sensitization (CAS) underlies atopic dermatitis (AD) and leads to various allergic symptoms, including food allergy and anaphylaxis. IL-13 expression by T follicular helper T (TFH) has been reported to be involved in generating high-affinity IgE antibodies and causing systemic anaphylaxis.1, 2 However, the mechanisms by which IL-13 triggers IgE-mediated allergic responses remain poorly defined. In the present study, we elucidate the role of IL-13 in the CAS-mediated mechanism by which high-affinity IgE antibodies are produced when the same allergen is introduced at a distal site in the secondary sensitization. The CAS model system using mice lacking the cell lineage-specific IL-13 receptor (IL-13R) demonstrated that dendritic cells (DCs), but not T or B cells, are critical in the high-affinity IgE-mediated anaphylactic response. The IL-13 signal in type 2 conventional…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
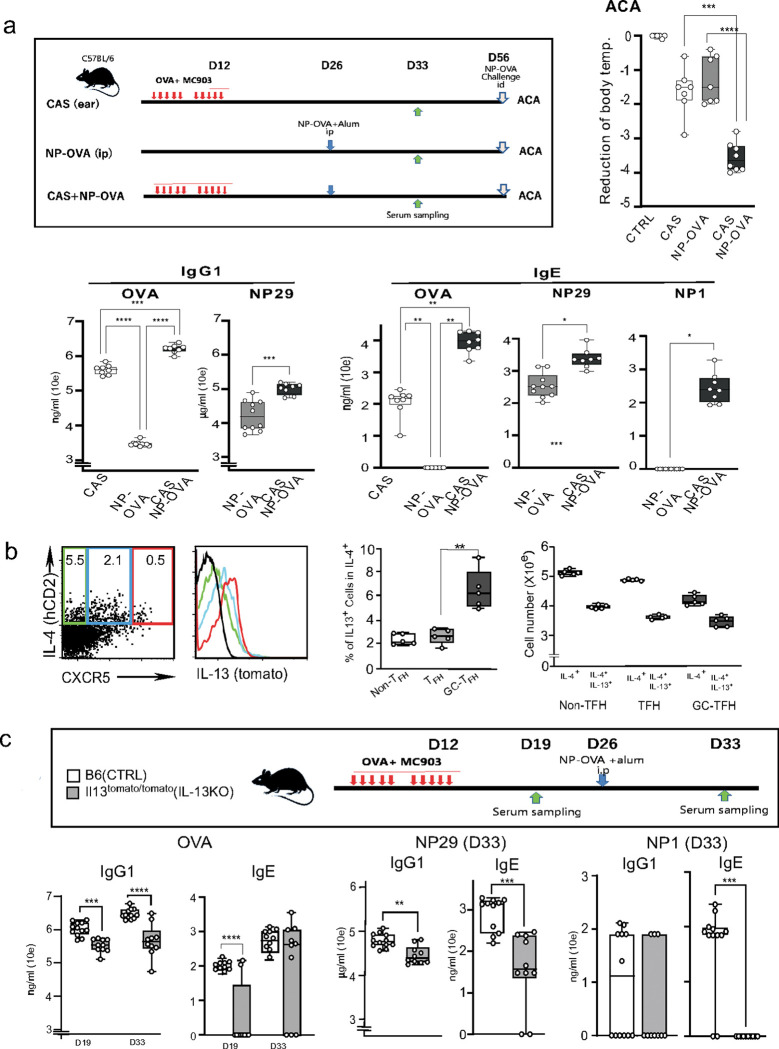 Figure 1
Figure 1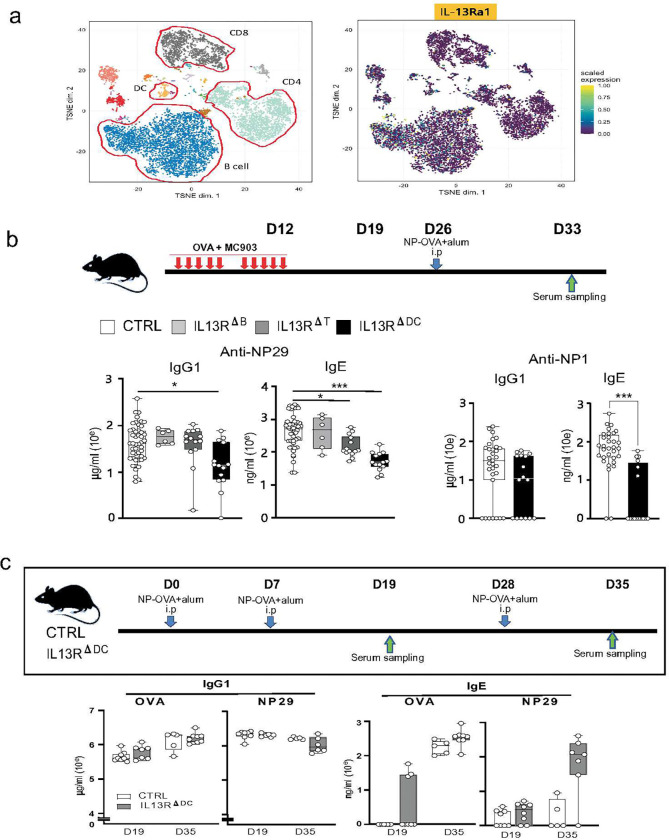 Figure 2
Figure 2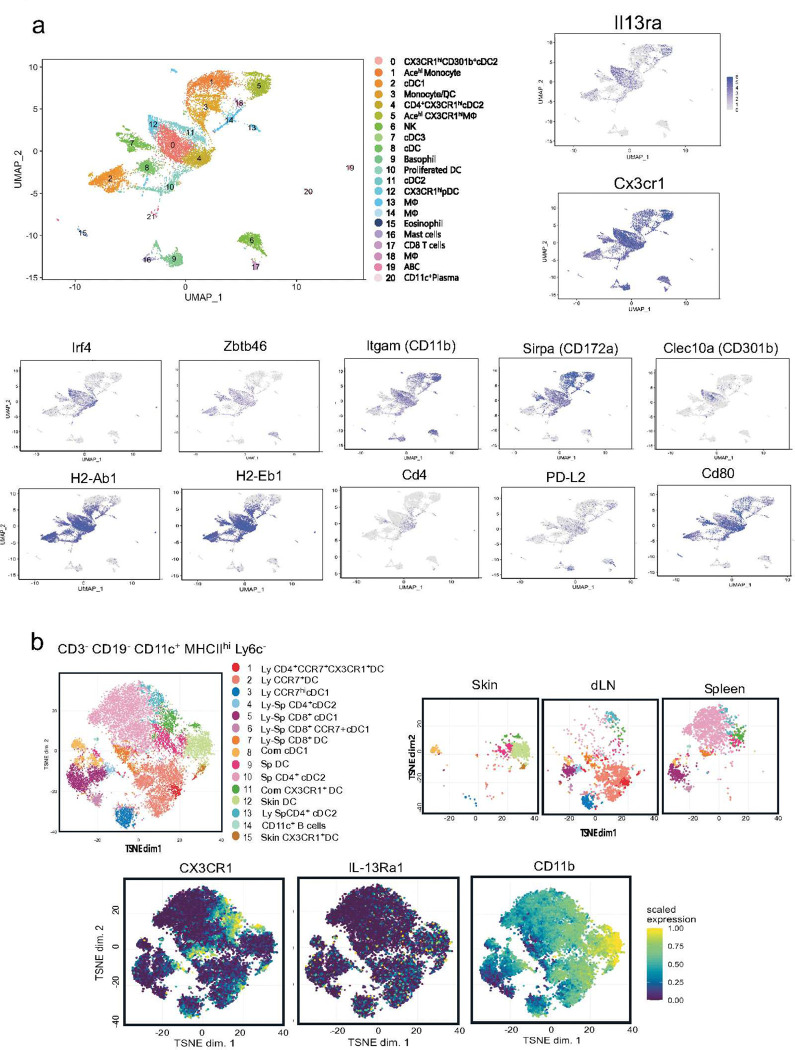 Figure 3
Figure 3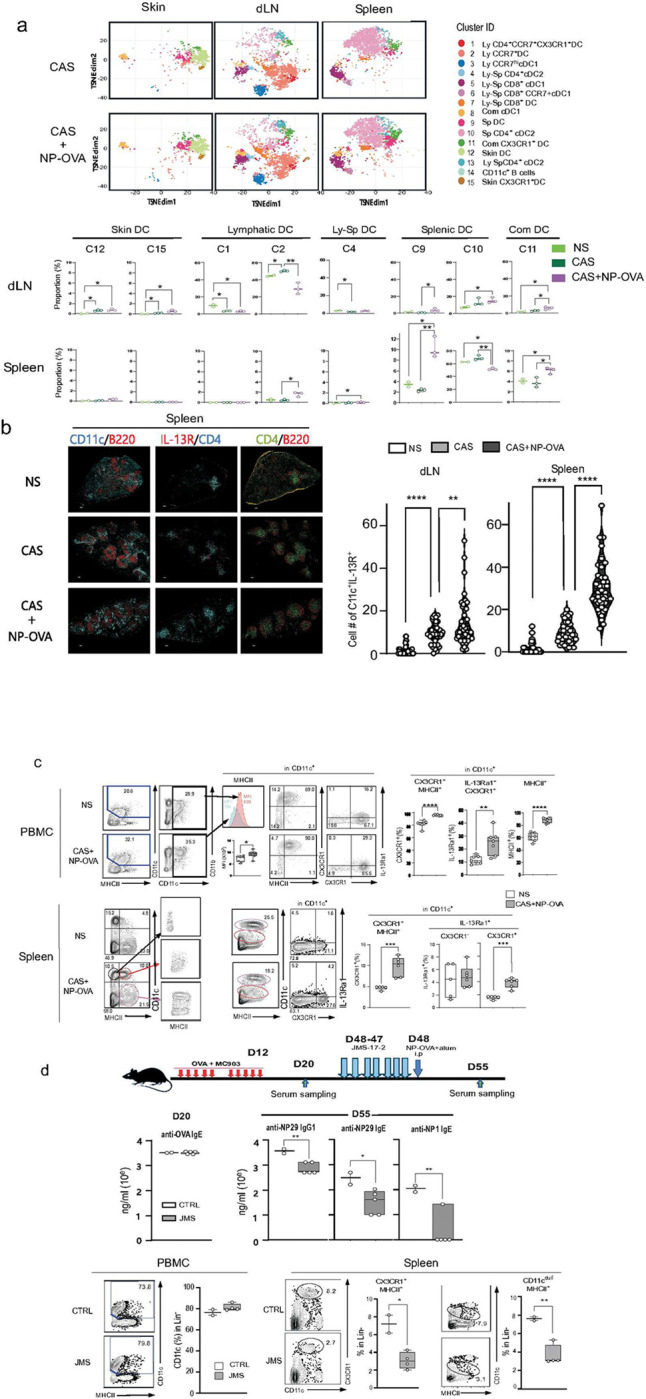 Figure 4
Figure 4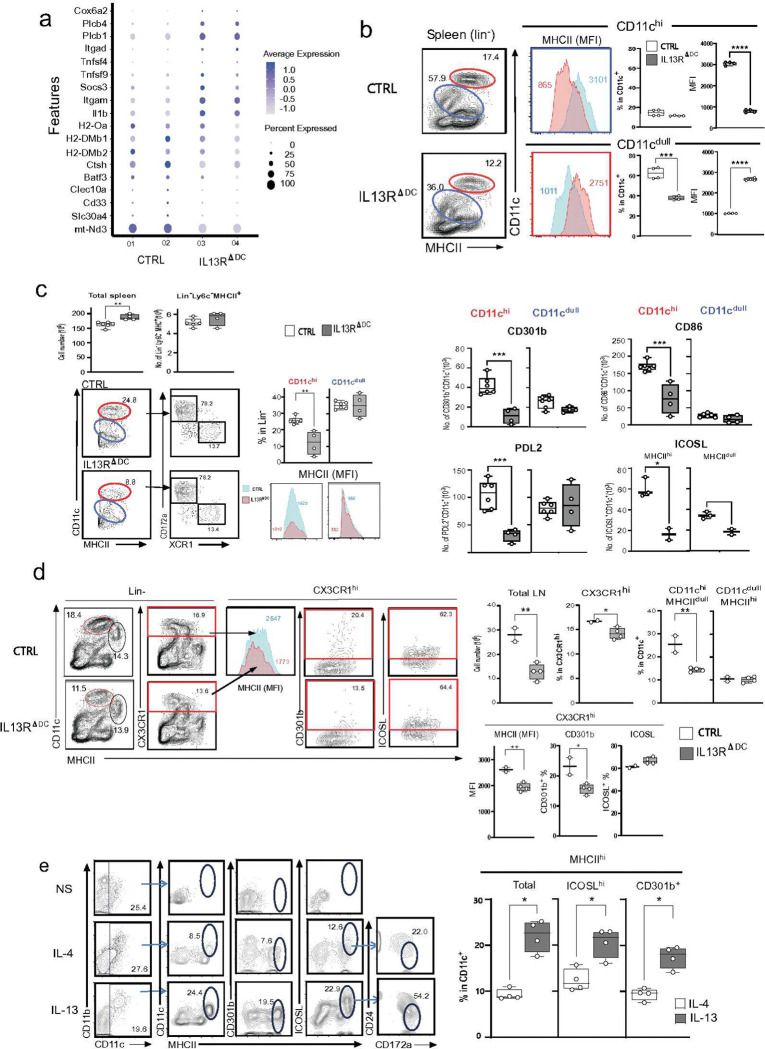 Figure 5
Figure 5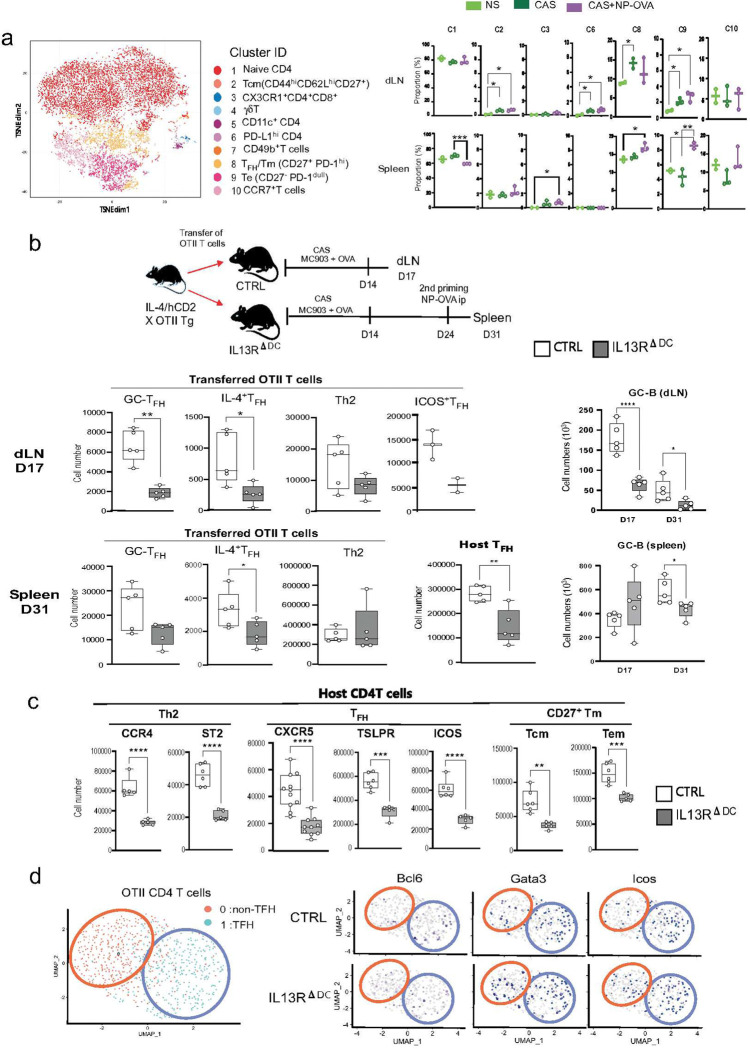 Figure 6
Figure 6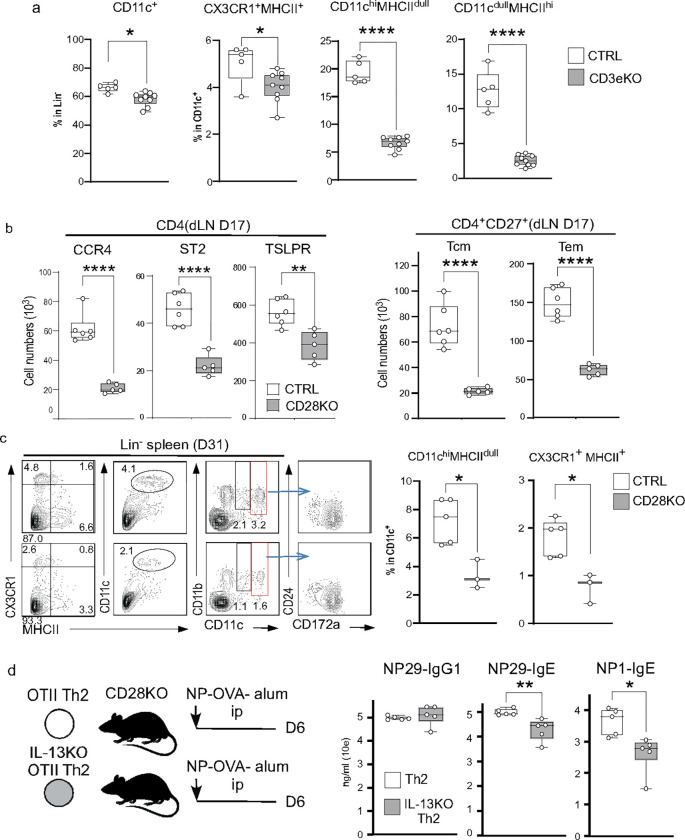 Figure 7
Figure 7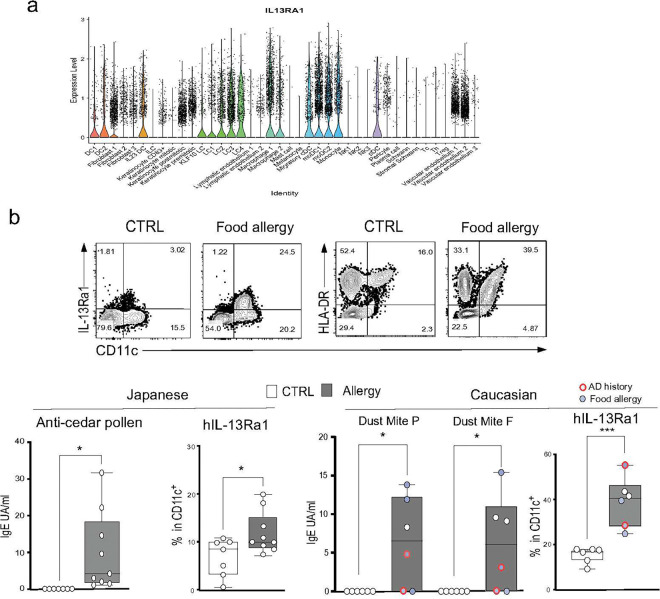 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDermatology and Skin Diseases · Food Allergy and Anaphylaxis Research · Urticaria and Related Conditions
Introduction
Allergy encompasses a complex array of diseases that combine innate and adaptive immunity, host genetic polymorphisms, and environmental triggers that collectively influence susceptibility to type 2 inflammation. Skin barrier dysfunction in the setting of atopic dermatitis (AD) leads to cutaneous allergen sensitization (CAS).^3^ The atopic march hypothesis proposes that such skin sensitization leads to the subsequent hypersensitivity to allergens at other barrier surfaces, resulting in asthma, food allergy, and its associated anaphylaxis.^4^ The damaged skin barrier in AD allows for the penetration of allergens, which are taken up by Langerhans and dendritic cells (DCs). Skin DCs then migrate to the local lymph nodes and present allergens to T cells, resulting in the generation of type 2 helper T (Th2) and T follicular helper T (T_FH_) cells, which produce the canonical type 2 cytokines interleukin (IL)-4 and IL-13. IL-13 is known to optimally promote the development of allergen-specific IgE, a defining feature of atopy.^5, 6, 7^ Thus, the secondary lymphoid organ represents a key anatomic site for allergen priming. However, the precise mechanisms that convert local allergen sensitization in the skin to systemic allergy remain poorly defined.
Epithelial cell-derived alarmin cytokines such as thymic stromal lymphopoietin (TSLP) and IL-33 act to promote type 2 responses, both innate and adaptive type 2 inflammation in the skin in response to barrier breach and are early initiators of allergen sensitization.^8, 9, 10, 11^ The vitamin D_3_ analog, calcipotriol (MC903), is known to be a potent inducer of TSLP, causing AD-like skin inflammation with barrier dysfunction.^8, 9^ We have also reported that TSLP receptor (TSLPR) expression on CD4 T cells was critical for the survival of Th2 cells.^12^ Analysis of Tslpr^flox/flox^ mice crossed to Zbtb4-cre or Cd4-cre lines has demonstrated that TSLPR signaling in T cells and DCs is vital for Th2-induced inflammation.^13^ In this context, TSLP-treated DCs promote Th2 differentiation in humans and mice by expressing CCL17 and CCL22.^14, 15^ On the other hand, it has been reported that MC903-induced TSLP expression causes T_FH_ development in mouse asthma models.^16^ Langerin^+^ and Langerin^−^ migratory skin DCs contribute to the TSLP-induced T_FH_ differentiation and GC responses, and antigen transport to draining LNs was mainly performed by Langerin^−^ DCs.^17^ Interestingly, TSLP is required for the T_FH_ and Th2 responses initiated by CAS, but not by dermacutaneous allergen sensitization.^18^ Therefore, TSLP expression in the skin caused by CAS is sufficient to induce local allergic reactions. However, how the CAS-mediated local responses lead to systemic IgE responses remains unclear.
In the lung, ILC2-derived IL-13 is necessary for attracting migratory DC2s (migDC2s) to the draining lymph nodes (dLNs) for Th2 cell priming by the upregulation of CCR7.^19, 20^ The migration of migDC2s into dLNs promotes the differentiation of Th2 cells, which express a specific receptor for IL-33. Activated Th2 cells relocalize back to the inflamed lung parenchyma and bronchoalveolar fluid to facilitate chronic airway responses.^21^ Following re-challenge with allergens or IL-33 injection, the Th2 cells attract eosinophils. Interestingly, these Th2-mediated secondary allergic responses were impaired in B cell-deficient μMT mice, in which T_FH_ cell development is defective, implying that T_FH_ cells could be precursors of Th2 cells.^21^ Additionally, it has been reported that steady-state production of IL-13 by skin ILC2s was necessary to facilitate the antigen-presenting ability of CD11b^mid^ migDC2s to favor Th2 differentiation.^22^ However, how IL-13 may contribute to the T_FH_-mediated humoral and memory responses to generate systemic IgE responses remains poorly defined.
A unique subset of IL-13-producing T_FH_ cells, called T_FH_13 cells, has been implicated in the production of anaphylactic high-affinity IgE Abs to inhaled allergens.^1^ We have also reported that Th2 cells could differentiate into GATA3^+^ T_FH_2 cells upon secondary antigen stimulation.2 Since activated B cells, including GC-B cells, express IL-13Rα1,^23, 24^ it was speculated that IL-13Rα1^hi^-expressing IgG1 GC-B cells may be the primary target of the T_FH_13-derived IL-13 to induce sequential class-switching to IgE.^1, 2^ On the other hand, IL-13 derived from ILC2s contributes to the IgE responses controlled by T_FH_ cells in response to peanut allergens via CD11c^+^ MHCII^hi^ DCs.^25^ Therefore, how IL-13 contributes to IgE responses and high-affinity IgE production remains unclear.
The IL-4/STAT6 axis is the critical pathway for IgE responses, as genetic deletion of Il4 or Stat6 eliminates both total and antigen-specific IgE responses.^26, 27^ The IL-4 derived from T_FH_ is critical for the production of allergen-specific IgE Abs in the GC response.^28^ In contrast, the role of IL-13 in IgE production remains controversial. Previous studies indicated that IL-13-deficient mice showed a selective reduction of IgE antibodies in an OVA immunization model but not in an infection model with the helminth Nippostrongylus.^29, 30, 31^ In contrast, the monoclonal Ab biologic lebrikizumab, which targets human IL-13, has shown a pivotal therapeutic effect in moderate to severe forms of AD as well as inhibiting IgE responses in adults.^32, 33^ Moreover, antigen exposure through the airways without adjuvant induced an IL-13-dependent T_FH_ response in the IgE response to peanut allergens.^16^ However, how IL-13 leads the allergen-specific IgE response independent of IL-4 is still unclear.
The IL-13 signal is transduced by the heterodimeric complex of the IL-13 receptor α chain 1 (IL-13Rα1) and IL-4Rα. The IL-13 signal results in activation of JAK1, JAK2, and tyrosine kinase 2 (TYK2) and subsequent phosphorylation of STAT3 and STAT6.^34, 35^ In human monocytes/macrophages, it was reported that IL-13 elicits Jak2-STAT3 and Jak1-STAT1 pathways.^36^ IL-4-activated STAT6 controls IgE class-switching in both human and mouse B cells, but there is no evidence to show a contribution of IL-13. These observations suggest that IL-13 controls IgE antibody (Ab) responses independently from IL-4 and that IL-13 binding to the cognate receptor provides an independent signal in different cell lineages.
In the present study, we show that cutaneous antigen sensitization via AD-like skin inflammation causes hyperactivation of Th2 cells in the dLNs, which subsequently triggers high-affinity IgE responses that cause anaphylaxis when the same T-dependent antigen is introduced at distal sites. Our series of analyses indicated that the specific activation of the cDC2 subset by IL-13 signal promoted the efficient generation of Th2 memory cells and T_FH2_ cells, which induce the production of allergen-specific IgE and high-affinity IgE antibodies. Therefore, IL-13-dependent cDC2 activation is a critical pathway controlling CAS-mediated allergic responses.
Results
CAS induces a high-affinity IgE response and IL-13-expressing TFH cells
Antigen sensitization in chronically damaged skin induced by topical application of MC903 leads to a break in tolerance, manifesting as a severe allergic reaction upon repriming with appropriate antigens. We applied topical MC903 daily along with the model antigen OVA, allowing proximity between the antigen and the damaged skin. The MC903-treated mice showed robust production of OVA-specific IgG1 and IgE (Fig.1a). To monitor the impact of CAS on the response to an inappropriate allergen by systemic antigen exposure, we further immunized the mice intraperitoneally with 4-hydroxy-3-nitrophenyl acetyl (NP)-OVA in alum adjuvant. In this case, the hapten NP is a primary antigen that B cells encounter for the first time. The NP-OVA immunization promoted about a 10-fold increase in OVA responses and 10- or 100-fold higher levels of anti-NP IgG1 or IgE antibodies than those seen following systemic immunization alone (Fig.1a). Moreover, the anti-NP IgE, but not IgG1, antibodies included high-affinity Abs recognizing NP1, but such high-affinity IgE did not appear with NP-OVA priming alone. These results indicated that the CAS-induced OVA-specific memory T cells could induce high-affinity anti-NP IgE Abs even upon primary B cell activation. Furthermore, the intradermal challenge with NP-OVA at the unlinked site caused severe anaphylaxis symptoms (Fig.1a), indicating that the high-affinity anti-NP IgE Abs were initiated by the CAS-induced OVA-specific memory T cells even when OVA was introduced at distal sites. The high-affinity IgE correlated with the severity of anaphylaxis.
IL-13 secreted by T_FH_13 cells plays a critical role in the high-affinity IgE response.^1, 2^ We first asked whether CAS induced the development of T_FH_13 cells. IL-4 and IL-13 double-reporter mice^37^ were treated with CAS and systemic immunization with NP-OVA, and we then compared IL-13 expression by CXCR5^−^ (Th2), CXCR5^mid^ (preT_FH_), and CXCR5^+^ T_FH_ cells among the IL-4^+^ CD4 T cells. The CXCR5^+^ T_FH_ cells were the most abundant population that highly expressed both IL-4 and IL-13 (Fig.1b). CAS-treated IL-13-deficient mice (IL-13KO), in which tomato reporter genes were knocked in at the Il13 locus on both alleles, ^37^ exhibited a significant reduction in the anti-NP29 IgG1 and IgE response (Fig.1c). The IL-13KO mice further showed a striking decrease in anti-NP1 IgE but not IgG1 Abs (Fig.1c). These results indicated that CAS played a role in the generation of T_FH_13 cells, which may control the induction of high-affinity IgE Abs.
IL-13 signaling in dendritic cells selectively contributes to the skin sensitization-mediated IgE responses.
T_FH_-derived IL-13 contributes to controlling sequential class-switching to IgE.^1, 38^ However, whether IL-13 acts directly on GC-B cells or indirectly via another cellular target is unclear. Therefore, we asked which immune cells mainly express the receptor for IL-13 (IL-13Ra1). We analyzed the expression of IL-13Ra1 by mass-cytometry (CyTOF) analysis of the cervical LN cells from mice treated with CAS and systemic immunization and found that it was dominant in B cells but was also significantly expressed by CD4 T and DCs (Fig.2a).
To further identify the primary target of the IL-13 signal in CAS and systemic immunization, we generated mice containing loxP-flanked Il13ra1 alleles (Il13ra1^f/f^) (S-Fig.1a&b) and crossed them to mice expressing Cre recombinase under the control of the T-cell-specific Cd4 promoter (Il13ra1^f/f^Cd4-Cre; referred to as Il13ra1^ΔT^), the B-cell-specific promoter of the gene encoding the BCR signaling subunit CD79A (Il13ra1Cd79a-Cre; referred to as Il13ra1^ΔB^) or the CD11c promoter (Il13ra1^f/f^Cd111c-Cre; referred to as Il13ra1^ΔDC^). After CAS and systemic immunization, we measured anti-NP IgG1 and IgE titers on day 33 (D33). We expected that B cells would be a direct target of IL-13 because B cells showed the most abundant expression of Il13ra1 transcript (S-Fig.1c). However, there was no significant reduction in the IgG1 and IgE responses to NP29 in the Il13ra1^ΔB^ mice (Fig.2b). The Il13ra1^ΔT^ mice showed a subtle but significant reduction in the IgE response to NP29 but no reduction in the IgG1 responses to NP29. In contrast, Il13ra1^ΔDC^ mice showed a marked reduction in the anti-NP29 IgG1 and IgE and the anti-NP1 IgE responses. The DC-specific deletion consistently showed selective loss of transcript and cell surface expression of IL-13Ra1 in CD11c DCs but not in T and B cells (S-Fig.1 c&d). Interestingly, the IL-13 signaling in DCs has a selective role in type 2 humoral responses, in which allergens primarily enter through the skin. The Il13ra1^ΔDC^ mice treated with systemic NP-OVA immunization without CAS treatment showed no reduction in the anti-OVA and anti-NP IgG1 after boosting (D35) (Fig.2c). Systemic immunization induced detectable levels of anti-OVA IgE and no or subtle anti-NP IgE antibodies in both control and Il13ra1^ΔDC^ mice. We also found a similar pattern following immunization with the SARS-CoV-2 spike protein (S-Fig.2). These findings suggest that DCs are the primary target of IL-13 in generating type 2 humoral responses, including high-affinity IgE antibodies. The IL-13-mediated type 2 humoral responses are selectively associated with initial allergen priming in the skin.
Skin sensitization promoted the accumulation of IL-13Ra1+ cDC2s in dLN and spleen.
The above results indicated a specific role of the IL-13 signal in DC for CAS-mediated allergic responses. Previous evidence suggested that IL-13 modified the DC character to activate Th2 responses.^(42,43)^ To identify the DC population responsible for the CAS-mediated allergic responses, we analyzed the expression of the Il13ra1 transcript by single-cell RNA sequencing (scRNAseq) of CD11c^+^ cells in the inflamed spleen after priming with the combination of CAS and systemic immunization (D33). An unsupervised Uniform Manifold Approximation and Projection (UMAP) algorithm identified 20 clusters, including six cDC clusters (Fig.3b). The cDCs were further subdivided into cDC1s (Irf8^+^ Xcr1^+^, Cd81^+^, Batf3^+^, GCsam^+^, Tcf4^+^), cDC2s (Sirpa^+^, S100a4^+^, Tcf4^+^, Klf4^+^, Irf4^+^) and cDC3s (Ccr7^+^, Nudt17^+^). Four clusters, C0, C4, and C11 contained cDC2-specific gene signatures. The Il13ra1 transcript was expressed in Ccr9^+^, Tcf4^+^, Siglec^+^pDC (C12), cDC2s (C0 and C11) and macrophages (C5). The Il13ra1 transcript was robustly expressed in C0 and C12. Several cDC2 transcriptional signatures, Zbtb46, Tcf4, Klf4, and Irf4 were abundant in C0. Interestingly, this cluster also expressed Cx3cr1. Furthermore, C0 also expressed several cDC2 marker genes, Sirpa (CD172a), Bst2, Clec10a (CD301b), and Pdcd1g 2 (PDL2) and highly expressed MHC2, Itgam (CD11b) and several activation markers, Cd80, Cd86, Icosl, Tnfsf (OX40L), Clec7a (Dectin-1), and Cd209a (CD-SIGN) (Fig.3b & S-Fig.3). These results indicate that in the inflamed spleen, IL-13Ra1 was predominantly expressed by CX3CR1^+^ cDC2s.
We further performed CyTOF analysis to examine cell surface expression of IL-13Ra1 on the MHCII^+^ CD11c^+^ population in independent tissues, skin, and cervical LNs, which are proximal to the initial priming site and the spleen, which is proximal to the secondary priming site. An unsupervised t-Distributed Stochastic Neighbor Embedding (t-SNE) algorithm^39^ identified 14 DC subsets among the three tissues we tested (Fig.3b). The DC populations in steady state (no stimuli; NS) were classified into tissue-specific subsets: the skin contained two dermal specific DCs, CX3CR1^+^ DC and CD11b^hi^ F4.80^+^DC,^22^ cervical LNs comprised four Lymphatic (Ly) subsets, CD4^+^CCR7^+^CX3CR1^+^DC (C1), CCR7^+^DC (C2), CD4^+^cDC2 (C4) and CCR7^hi^ cDC1 (C3), spleen composed of two splenic subsets, splenic DC (Sp DC, C9) and CD4^+^ cDC2 (C10) (Fig.3b). There were six shared populations: three CD8^+^cDC1 (C5, 6 and 7) and C4 were shared by LN and spleen, cDC1 and CX3CR1^+^ DC were found in all three tissues (Com cDC1; C8, Com CX3CR1^+^ DC; C11). Cell surface expression of IL-13Ra1 was found in two skin DC subsets (C12 and C15), C1, C2, C4, C9, C10, and C11. C1, C11, and C15 highly expressed CX3CR1, a membrane-bound receptor for the fractalkine CX3CL1, widely expressed by monocytes, macrophages, and monocyte-derived DCs.^40, 41, 42, 43^ These results suggested that CX3CR1^+^DCs were migratory, contributing to antigen delivery.
The above results suggested that the in vivo dynamism of IL-13Ra1^+^DCs would be the critical link between the flamed dLNs and the spleen. To examine this possibility, we performed a comprehensive CyTOF analysis to study the tissue dynamism of 14 distinct DC subsets in NS, local skin priming (CAS), and the combination of CAS and systemic immunization (CAS+NP-OVA) (Fig.3b & 4a). Following CAS treatment, the cell number in the cervical LNs increased at D19, while the proportions of C1 and C4 deceased. In contrast, the major subset in the dLN, C2, showed a significant increase (Fig.4a). In this condition, skin DC C12 and C15 - the subsets expressing IL-13Ra1 in the steady state - increased in the D19 dLNs, suggesting that the IL-13R+ skin DC migrate into dLNs around the CAS site. Systemic immunization with NP-OVA increased C9 and C11 in the spleen, where the allergen entered. The major subset in the spleen, C10, was significantly decreased, while the C9, C10 and C11 subsets increased in dLNs (Fig.4a). In addition, IL-13Ra1 was detected in C2 and C1 of the flamed dLNs, and C11 and C9 of the flamed spleen (Fig.3b). These results indicated that IL-13Ra1 is co-expressed with CX3CR1 in the neighboring SLO of the allergen entry.
Consistently, immunohistological staining indicated the emergence of the IL-13R^+^ DCs in the dLN and the spleen following CAS+NP-OVA treatment. The number of IL-13Ra1^+^ DCs significantly increased in the CAS+NP-OVA group compared to unprimed mice, with a marked increase compared to CAS alone (Fig.4b). The IL-13R DCs were predominantly localized in the T cell zone, the T-B border area, and the interfollicular regions (Fig.4b, S-Fig.5a). Notably, IL-13Ra1 expression overlapped well with CX3CR1 and CD301b (S-Fig.5a&b), which is consistent with the scRNAseq results (Fig.3a). Taken together, these results indicated that IL-13R DCs preferentially migrated and localized in the T-B border area and the interfollicular regions where they contribute to T cell activation in the spleen.
Interestingly, CX3CR1^+^ DCs were abundant in peripheral blood, and approximately 80% of DCs (CD11c^dull^ CD11b^hi^) were CX3CR1^hi^ even under a steady state. CyTOF analysis of peripheral blood mononuclear cells (PBMC) indicated that this CX3CR1^hi^ peripheral blood DCs (PBDC) corresponds to C11 in Fig.4a (S-Fig.6). Following CAS+NP-OVA treatment, the CX3CR1^hi^ PBDC increased to over 90% with the increased expression of IL-13Ra1^+^DCs and MHCII (Fig.4c). A similar increase in CX3CR1^+^MHC^+^DCs was also observed in the spleen after the CAS+NP-OVA treatment. These expanded DCs exhibited similar phenotypes to PBDCs, CX3CR1^hi^ CD11c^dull^ CD11b^hi^. IL-13Ra1 expression was significantly increased in splenic CX3CR1^hi^ DCs, with no significant change in CX3CR1^lo^ DCs due to more considerable deviation in the steady state (Fig.4c).
Previous reports have shown that CX3CR1^+^DCs migrate to inflamed SLOs through CX3CR1-CX3CL1 interactions.^44^ Therefore, we hypothesized that the CX3CR1^+^ PBDC might play a role in delivering NP-OVA allergen from peripheral blood to the spleen in systemic immunization. To test this possibility, we used the CX3CR1 antagonist, JMS-17–2 (TargetMol), to block the CX3CR1-CX3CL1 interactions. The CAS-treated mice with significant anti-OVA IgE Ab titers were treated with JMS-17–2 (JMS) prior to the systemic NP-OVA priming (Fig.4d). The JMS treatment significantly inhibited anti-NP IgG1 and IgE responses and decreased the proportion of CX3CR1^+^DCs in the inflamed spleen, while slightly increasing CX3CR1^hi^DCs in PBMC (Fig.4d). These results indicated that the CX3CR1+ DCs were required for allergen delivery to the SLO around the secondary priming site.
The IL-13 signal controlled the expression of MHCII and CD301b on DCs.
The above results indicated that CX3CR1 plays a role in allergen delivery via CX3CR1-CX3CL1 interactions. However, how the IL-13 signal in DCs leads to high-affinity IgE Ab production remains still unclear. Thus, we compared the scRNAseq data between the control- and the IL-13R^ΔDC^- derived splenic DCs. Gene expression indicated that the IL-13R^ΔDC^ DCs had decreased expression of MHC-related genes (H2-Oa and H2-DMb1 and -DMb2) and cDC2 signature genes, batf3, and Clec10a (CD301b) (Fig.5a). Therefore, the IL-13 signal may regulate the expression of MHCII in DCs. There are three DC populations, CD11c^hi^ MHCII^+^, CD11c^dull^ MHCII^lo^, and CD11c^lo^ MHCII^+^ DCs, in the spleen (Fig.5b). When we compared the MHCII expression between CD11c^hi^ (red) and CD11c^dull&lo^ (blue) DCs, the unprimed IL-13R^ΔDC^ mice exhibited significantly decreased mean fluorescence intensity (MFI) of MHCII on the CD11c^hi^ DCs but not on the CD11c^dull&lo^ DCs (Fig.5b). These results indicated that the CD11c-cre deletion in the IL-13R^ΔDC^ mice affected on the MHCII expression level on CD11c^hi^ DCs
The effect of the IL-13 signal was also observed in the splenic DCs after treatment with CAS+NP-OVA (Fig.5c S-Fig.7). The IL-13Ra1^ΔDC^ mice showed a significant reduction in the CD11c^hi^ DCs but not in the CD11c^dull&lo^ DCs despite no change in the MHCII MFI. As shown in Fig.4c, the fraction of CD11c^dull&lo^ DCs contained PBDC-derived CX3CR1^+^DC, and consistently, the IL-13Ra1^ΔDC^ PBDC showed no change in the proportion of the MHC^+^DCs, which displayed CD11c^dull^ (S-Fig.8). These results indicated that IL-13 signal was defective only in CD11c^hi^DCs of the IL-13Ra1^ΔDC^ mice. These defective DCs predominantly expressed CD172a, the marker of cCD2s, and co-expressed costimulatory molecules, such as CD301b, PDL2, CD86, CXCR5, and ICOSL.^45^ The expression of CD301b, PDL2, and CD86 was increased in control CD11c^hi^ DCs, while the cell number and the proportion of the CD11c^hi^DCs, but not in the CD11c^dull^DCs, expressing these costimulation molecules were significantly reduced in IL-13R^ΔDC^ mice (Fig.5c S-Fig.7). Previous literature reported that CD301b is a marker for the cDC2s directing Th2 differentiation.^46^ Therefore, we speculated that the role of the IL-13 signal in DCs is to control cDC2 functions, thereby promoting efficient type 2 humoral responses.
Next, we investigated the role of IL-13 signaling in DC function in the dLN following the CAS treatment. In the cervical LNs near the CAS-treated site, the control mice exhibited two major populations, CD11c^hi^MHC^dull^ DCs, and CD11c^dull^MHC^hi^ DCs (Fig.5d). CX3CR1 was highly expressed in CD11c^hi^ DCs but not in CD11c^dull^ DCs. The IL-13Ra1^ΔDC^ mice showed a marked reduction of the CD11c^hi^ DCs. The IL-13Ra1^ΔDC^ mice also exhibited reduced expression of MHCII, CD301b, PD-L2, and CD86 in CD11c^hi^MHC^dull^ DCs but not in CD11c^dull^MHC^hi^ DCs (Fig.5d, S-Fig.8). Therefore, it is reasonable to conclude that the IL-13 signal contributes to the accumulation of migratory CX3CR1^+^DCs in the LN localized close to the skin sensitization site.
Previous reports indicated that CX3CR1 promotes the development of myeloid precursors into DCs under steady-state conditions.^47^ Thus, it is likely that CX3CR1^+^ PBDCs would be a possible precursor of cDC2s.To investigate this possibility, we isolated CX3CR1^+^PBDCs and cultured them with IL-4 or IL-13 (Fig.5d, S-Fig.6). IL-4 enhanced the number of MHC^+^DCs, while IL-13 expanded more MHCII^hi^DCs compared to IL-4. Moreover, IL-13 significantly increased the expression of ICOSL by CD301b^+^MHCII^hi^DCs (Fig.5d). These MHCII^hi^ DCs contained CD24^dull^CD172a^+^ cells, indicating that most of the cells were cDC2s. Taken together, these in vivo and in vitro data suggest that the IL-13 signal in DCs promoted the maturation of cDC2s, directing Th2 differentiation by enhancing the expression of MHCII, CD301b, CD86, PD-L2, and ICOSL. The increased expression of MHCII and CD86 in CD301b^+^ cDC2s was thought to enhance antigen-presenting ability, promoting T cell activation in type 2 humoral immunity.
The IL-13 signal in cDC2s impacts the generation of IL-4-producing TFH cells and memory T cells.
The results in Fig.5d demonstrated that the IL-13 signal affects the ability of CD11c^hi^cDC2s to function as antigen-presenting cells even in the cervical LN after CAS treatment. Moreover, the anti-OVA IgG1 and IgE responses induced by CAS treatment alone were significantly lower in the IL-13R^ΔDC^ mice than in the control (S-Fig.9). Therefore, we speculated that the defect of the IL-13 signal affects primary T cell activation. To understand which CD4 T cell subsets are activated in the dLNs and the spleen of mice treated with CAS and CAS+NP-OVA, we examined changes in the CD4 T cell subsets by CyTOF analysis. The t-SNE projection indicated T_FH_ /memory T (Tm)(CD27^+^PD-1^hi^), central Tm (Tcm) (CD44^hi^CD62L^hi^CD27^+^), and PD-L1^hi^ CD4 T cells subsets were increased in the CAS treated dLNs (Fig.6a, S-Fig.10). On the other hand, the CAS+NP-OVA treated spleen exhibited an increase of T_FH_ /Tm, effector T (Te)(CD27^−^PD-1^dull^), and CCR7^+^T subsets. These results indicated that skin sensitization by CAS treatment actively enriches T_FH_ and memory CD4 T cells.
To confirm the role of the IL-13 signal in primary and secondary CD4 T cell activation, naïve CD4 T cells obtained from the OTII mice crossed with hCD2 IL-4 reporter (IL-4/hCD2) mice were transferred into control or IL-13R^ΔDC^ mice to track the expression of IL-4/hCD2 in Th2 (CXCR5^−^) and T_FH_ (CXCR5^+^) cells (Fig.6b). The number of IL-4^+^ T_FH_ cells and Th2 cells decreased in the dLN following CAS treatment, and a similar reduction of IL-4^+^ T_FH_ cells was also found in the spleen and dLN after the secondary priming on D31. Along with the decline in T_FH_ cells, the IL-13R^ΔDC^ recipient mice showed a significant reduction in the number of GL-7^+^GC-B cells in the dLNs on D17 and D31 and in the spleen on D31 (Fig.6b). The reductions were prominent among host Th2 and T_FH_ cells compared to the transferred OTII T cells (Fig.6c, S-Fig.11). CAS treatment upregulated several Th2 markers, CCR4 and ST2/IL-33R, and the T_FH_ markers, ICOS, and IL-13R^ΔDC^ mice showed prominent reductions of these makers. Previous studies have shown that MC903-induced TSLP expression supports Th2 and T_FH_ differentiation in several mouse models.^16, 48^ Consistent with these findings, IL-13R^ΔDC^ mice had a prominent reduction of TSLPR expression in the host CD4 T cells (Fig.6c). We also observed a reduction in CD27^+^ central memory (Tcm)(CD62L^hi^CD44^hi^) and effector memory (Tem) (CD62L^−^CD44^hi^) cells (Fig.6c). Furthermore, scRNAseq analysis indicated the transferred OVA-specific OTII T cells differentiated into two major clusters: C0 and C1 on D31 of the spleen (Fig.6d). The C1 island contained T_FH_ signature genes (Bcl6, Cxcr5, and Icos), while the C0 island had non-T_FH_ signature genes (Ccr7, S1pr1 and Gata3). Interestingly, the expression of the T_H_2 master regulator Gata3 was more abundant in C1 than C0, indicating that the transferred OTII T cells had differentiated into T_FH_ cells expressing GATA-3 and IL-13, corresponding to T_FH_2 or T_FH_13 cells (Fig.6d).^1, 2^ The distribution pattern of the Bcl6-, Gata3-, and Icos-expressing OTII T cells was less abundant in C1 of IL-13R^ΔDC^ mice compared to the control mice. IL-13 expressing T_FH_2 cells were not present in the IL-13R^ΔDC^ mice (S-Fig.12). These results indicate that the DC-derived IL-13 signal is critical for generating the T_FH_2 cells co-expressing IL-4 and IL-13, both of which are required for generating high-affinity IgE Abs.
T cell-derived IL-13 controls the fine-tuning antigen-presenting ability of cDC2s.
Previous literature has reported that skin ILC2-derived IL-13 in steady-state facilitates the antigen-presenting ability of CD11b^dull^migDC2s.^22^ Indeed, a decrease in MHCII expression was observed in the splenic DCs of IL-13Ra1^ΔDC^ mice (Fig.5b). To investigate whether the ILC2-derived IL-13 contributes to the migration of DCs into dLNs in the CAS-induced mouse, we treated Cd3e deficient mice with CAS and on D17 assessed the number of CD11c^hi^MHC^dull^ DC subset cells in the dLN, which contained a large number of CX3CR1^+^DCs (Fig.7a, S-Fig.13). The loss of T cells induced a striking reduction in the attraction of two major mature DC subsets, CD11c^hi^MHC^dull^ DC and CD11c^dull^MHC^hi^ DCs even though the total number of DC in the dLN was unchanged. These results suggested that T cell-derived IL-13 might be required for the initial maturation of the DCs localizing in the dLN near the skin sensitization site.
Next, we hypothesized that the OVA-specific memory Th2 cells generated by CAS treatment could become the IL-13 source in the secondary response. To test this hypothesis, we used CD28-deficient mice, which were impaired in memory formation due to the lack of initial T cell activation (Fig.7b). CAS-treated CD28-deficient mice exhibited a striking reduction in the number of CD4 T cells expressing pathogenic markers, CCR4, ST2, and the number of Tcm and Tem cells was also markedly reduced in the dLNs on D17 (Fig.7b). Under this condition where Tcm and Tem cells were less dominant, the spleen of the CD28-deficient mice additionally immunized with NP-OVA on D31 showed significantly fewer CX3CR1^+^DCs and CD11c^hi^MHCII^dull^ DCs, which also expressed the cDC2 marker, CD172a (Fig.7c). Therefore, these results suggest that the OVA-specific Tm cells could be the primary source of IL-13 required for the maturation of CD11c^hi^MHCII^dull^cDC2 in the spleen after systemic immunization.
We recently reported that Th2 cells further differentiate into IL-13-producing T_FH_2 cells after secondary antigen stimulation.^2^ Similar T_FH_2 cells were also generated in the spleen of mice treated with CAS+NP-OVA (Fig.1b), suggesting that memory Th2 cells could be a source of the IL-13 required for cDC2 maturation regulating T_FH_2 generation. Therefore, we investigated the role of IL-13 from Th2 cells in the IgG1 and IgE responses using adoptive transfer of ex vivo generated Th2 cells (Fig.7d). The transfer of OTII-derived Th2 cells lacking IL-13 resulted in significantly lower titers of anti-NP IgE and high-affinity IgE, but not IgG1, than in mice transferred with IL13-sufficient Th2 cells (Fig.7e). These results indicated that memory Th2 cells-derived IL-13 controlled cDC2 maturation required for the IgE and high-affinity IgE responses.
Human L-13Ra1 on PBDC is a marker of AD history
Fig.4c indicated that CX3CR1^+^PBDCs increased the expression of IL-13Ra1 and MHCII following CAS+NP-OVA treatment, which mimics secondary allergen sensitization in human AD patients. Therefore, we assumed that IL-13Ra1 and MHCII expression on PBDCs would be markers for the CAS-mediated activation. The expression of IL-13Ra1 in skin DCs has also been demonstrated in a recent large-scale cohort study, and IL-13Ra1 has been shown to be expressed constitutively by migratory cDC of healthy human skin (Fig.8a).^49^ We therefore conducted further analysis to investigate the correlation between IL-13Ra1 and MHCII in PBDCs newly isolated from human allergy patients. The Japanese subjects included nine with allergic rhinitis to cedar pollen (CP) who had IgE antibodies specific for CP, and six negative controls for analysis during the pollen non-dispersal period. The Caucasian subjects included six with IgE antibodies specific for food and dust mite allergens and six negative controls. This group also included two with active AD and four with food allergies. We found a significant increase in the percentage of IL-13Ra1^+^DCs in both Japanese and Caucasians (Fig.8b), although the trend toward increased expression of IL-13Ra1 was more prominent in the Caucasian group. All six individuals also showed an increased number of MHCII^hi^ (HLA-DR) DCs (Fig.8b). Therefore, consistent with the mouse CAS model, the expression of IL-13Ra1 and MHCII by circulating DCs correlated with the CAS and AD history. These results demonstrated that the IL-13Ra1^+^DCs may circulate in the bloodstream of individuals with AD history. Therefore, IL-13Ra1 and MHCII expression on PBDCs should be good markers for AD history and predict susceptibility to future allergic march.
Discussion
The present work indicated that CAS confers a specific type 2 microenvironment that promotes high-affinity IgE responses via the IL-13-mediated activation of cDC2s. In the primary CAS response, the IL-13 signal is critical in promoting Th2 memory and T_FH_ responses by accumulating CX3CR1^+^ migratory DCs in dLNs. In the secondary response, CX3CR1^+^ PMDCs were necessary for allergen delivery into distal lymphoid tissue to induce IL-13 production by memory Th2 cells. The Th2-derived IL-13 plays a role in expanding the MHCII^+^cDC2s to confer better antigen presentation capability. The IL-13 signals also enhanced the expression of CD301b, PD-L2, and CD86 by the MHCII^+^cDC2s, accelerating the differentiation of T_FH_2 cells and subsequently enhancing the GC-dependent allergen-specific high-affinity IgE response. Therefore, a serial combination of IL-13 signals and CX3CR1 expression by the DCs are central in connecting skin and distal lymphoid organs in a continuum.
The first studies of IL-13 loss-of-function mice indicated a subtle effect on Th2 cytokines and a robust impact on IgE but not on other isotype responses.^30^ Loss-of-function and blocking studies also suggest that IL-13 signaling appears involved in IgE responses induced primarily by antigen exposure through the airways and skin but not in systemic responses.^29, 30, 31^ Previous reports proposed that T_FH_13-derived IL-13 independently controls high-affinity IgE antibody production via sequential class-switching in the GC-B cells.^38^ However, the present study demonstrates that IL-13Ra1 expressing DCs, especially cDC2s, are a significant target in the IgE responses against the allergen. Interestingly, the IL-13-dependent pathway was selectively active only when the initial allergen was introduced from damaged skin.
It has been reported that IL-13 is critical for the functional modification of dermal DCs to promote Th2 generation and attraction in allergen protease-induced asthmatic inflammation.^19, 20, 21, 50^ A previous study suggested that the skin resident CD11b^mid^ cDC2s may be a target of IL-13 signaling at steady state.^22^ In this case, homeostatic IL-13 from dermal ILC2s regulates the population size of CD11b^mid^ cDC2s to promote Th2 differentiation. However, the CyTOF data in Fig.3b indicated that CD11b^hi^DCs (C12) would be skin resident DCs, while the CD11b^mid^ skin DCs (C15) highly expressed the fractalkine receptor CX3CR1 responsible for attracting DCs to the lymphatic endothelium to establish type 2 microenvironments in the dLN.^51, 52^ Since the CX3CR1^+^ DCs were abundant in PBMC (S-Fig.6), they were the population circulating in the blood and lymphatic vessels. Interestingly, both skin DC populations expressed IL13Ra1 and significantly increased in the cervical LN after CAS (Fig.4a). The CX3CR1^+^ DCs were CD11c^hi^ in the dLNs around the CAS site. They were significantly reduced in the absence of the IL-13 signal (Fig.5d), suggesting antigen delivery to dLNs by the skin DCs was IL-13-dependent under the CAS condition. This observation was consistent with the global view of the immune cell responses to 86 cytokines, suggesting that cDCs and migDC are primary functional targets of IL-13 in mouse LNs in vivo.^53^ The CD301b^+^ DCs within the subepithelial regions in the skin are reported to be responsible for transporting cutaneous antigens to the dLN.^46^ Our in vivo results demonstrated that loss of the IL-13 signal in the DCs reduced the number of CD301b^+^DCs, indicating the contribution of the IL-13 signal to the expression of CD301b by the CX3CR1^+^migDCs. Previous reports indicated that IL-13 enhances Th2 differentiation via the interaction between antigen-loaded migDC2s and CD4^+^ T cells.^54^ Recently, “macro-clustering” of CD301b^+^ migDC2s has been reported to promote and specialize Th2 cell development.^55^ Therefore, the IL-13 signal contributes significantly to the accumulation of CX3CR1^+^ DCs and expression of MHCII and CD301b, directing to a robust Th2 response, and then some of the Th2 cells could remain and circulate as memory Th2 cells.
IL-13 signaling in DCs is also associated with high-affinity IgE responses in secondary systemic responses. However, unlike the CAS-mediated response, the target of IL-13 signaling would be CX3CR1^−^cDC2s, which constitute a significant CD11c^hi^ population in the spleen. Because they also express IL-13Ra, IL-13 influenced the CX3CR1^−^cDC2s to increase the expression of MHCII, CD301b, PD-L2, and CD86 (Fig.4c & Fig.5c). This is consistent with the in vitro culture. IL-13 selectively promoted the maturation process of cDC2, which expressed high levels of MHCII, CD301b, and ICOSL (Fig.5d). The effect of IL-13 was more pronounced than that of IL-4. These results suggest that IL-13 signaling is a potent driver of cDC2s to promote type 2 T cell responses. This observation raises the possibility that some cDC2s are derived from CX3CR1^+^PBDCs. Therefore, in the secondary responses, the IL-13 signal acted mainly on the cDC2 population, accelerating the expression of MHCII and several costimulatory molecules that drive the T_FH_2 response. Moreover, IL-13 signaling was also required to modify the immunological characteristics of patrolling CX3CR1^+^PBDCs into cDC2s. Another important mechanism in secondary responses was the role of CX3CR1^+^DC, which is required for antigen delivery to Th2 memory cells in the spleen. The CX3CR1-CX3CL1 interaction may allow the CX3CR1^+^PBDCs to enter the inflamed spleen, where the blood vessel endothelium expresses CX3CL1.^(51,54)^ A previous publication showed that CX3CL1-neutralizing antibodies inhibit the migration of allergen-loaded DCs to lymph nodes and inflammatory sites.^(52)^ Blocking CX3CR1^+^ DC entry into the spleen with the CX3CR1 antagonist JMS-17–2 abolished anti-NP IgG1 and IgE responses after systemic immunization with NP-OVA (Fig. 4d), supporting the idea that CX3CR1 expression may be a critical mechanism in controlling the influx of patrolling CX3CR1^+^ DCs into the inflamed spleen.
In secondary responses, memory Th2 cells would be the initial source of IL-13, driving activation of cDC2 in the spleen because in vivo transfer of OVA-specific Th2 cells generated high-affinity anti-NP IgE Abs after NP-OVA second priming, whereas the transfer of IL-13-deficient Th2 impaired these IgE responses (Fig.7d). CD28-deficient mice showed a lack of IgE responses along with a significant reduction of cDC2s (CD11c^hi^ MHCII^dull^) in the spleen (Fig.7c). These results suggest that memory Th2-derived IL-13 is essential for the activation of cDC2s required for the generation of allergen-specific high-affinity IgE Abs. More interestingly, CAS+NP-OVA selectively generated the IL-13-expressing T_FH_2 cells (Fig.1b). However, the OTII T cells transferred into IL-13Ra1^ΔDC^ recipient mice had less IL-13 expression in the T_FH_ cluster (S-Fig.10). Therefore, in a later phase, the Th2-derived T_FH_2 would be an alternative source of IL-13 to accelerate MHCII expression on cDC2s in a paracrine manner.^2^ On the other hand, the source of IL-13 involved in the initial DC activation of the dLN immediately after CAS treatment appears to be mechanistically different. In CD3ε-deficient mice, the accumulation of DCs in the dLN was drastically reduced (Fig.7a), suggesting that T cell-mediated IL-13 may also be required for traffic between the skin and the LN, but how the initial trigger of Th2 is introduced remains unclear. Therefore, the exact mechanism by which Th2-derived IL-13 regulates DC migration requires further investigation.
The CAS concept has also been found to be the case in a human cohort study showing that CAS in the context of AD, but not in the non-AD context, increases the risk of asthma as part of the atopic march.^(64–66)^ Consistently, type 2 inflammation increased the number of IL-13R+ PBDCs in humans and mice (Fig.4c & Fig.8b). Thus, the increased expression of MHCII and IL-13Ra1 are excellent markers to trace whether initial antigen priming is introduced via the skin. These results suggest that initial skin antigen sensitization efficiently dictates type 2 immune properties in distal lymphoid organs via circulating memory Th2 cells and the attraction of the IL-13R^+^ DCs.
In conclusion, the present study proposes the importance of IL-13 signaling in connecting the skin barrier and the secondary lymphoid organs. In the context of skin sensitization, dermal DCs and/or CX3CR1 DCs play a role in allergen delivery to LNs and spleen, and IL-13 has a selective role in providing the activation signal for cDC2s to increase antigen-presenting capacity and promote robust IgE responses via memory Th2 and T_FH_2 cells. IL-13-dependent cDC2 activation induced by increased allergen penetration into the skin is tightly linked to allergen-specific pathogenic IgE responses. Therefore, IL-13R DCs could be reasonable therapeutic targets in IgE-mediated allergic responses and in controlling the atopic march.
Material and Methods
Mice
The mouse strains used in these experiments, C57BL/6J, OTII, and IL-4-hCD2Bac Tg mice, are described in previous reports.^28^ IL-13-tomato mice were kindly provided by Andrew Mackenzie (MRC Laboratory of Molecular Biology).^37^ Icos^−/−^ mice were kindly provided by Japan Tobacco Inc. (Tokyo, Japan).^56^ Cd4-cre and Cd79a-cre mice are described in previous reports,^57^ and Cd11c-cre mice were a gift from Boris Reizis (Columbia University).^58^ Il13ra1 floxed mice were generated using the CRISPR/Cas9 system (Cyagen Santa Clara, CA. USA). Targeting contracts using a self-deletion anchor-containing neo cassette and the primers used for construction and genotyping are described in S-Fig.1 and S-Table 1. Targeted ES cell clones were injected into C57BL/6 albino embryos, which were then re-implanted into CD-1 pseudo-pregnant females. Founders were identified by their coat color and germline transmission was confirmed by breeding with C57BL/6J females.
All experiments were conducted with the approval of the Tokyo University of Science and RIKEN IMS Institutional Animal Care and Use Committees. Animals were housed on a standard 12:12 hour light-dark cycle with free access to food and water. All mice used in this study were adults (8–12 weeks) and were maintained under specific pathogen-free conditions. Mice were either randomly assigned to treatment groups or assigned to groups based on genotype. Experiments were performed on independent cohorts of male and female mice. No differences between sexes were observed and no analyses of the influence of sex were performed.
Human subjects and isolation of cells
Nonallergic healthy volunteers and volunteers with seasonal allergy symptoms around the time of seasonal pollen dispersal were recruited at the Department of Pulmonary Medicine, Kagoshima University. Healthy subjects with no nasal clinical history and no IgE for specific Japanese cedar pollen, cypress pollen, house dust mites, orchard grass pollen, or ragweed pollen were included as healthy controls. Subjects in the Japanese cedar pollen-induced allergic rhinitis group had typical nasal symptoms during the cedar pollen season and elevated IgE specific for Japanese cedar pollen (≥0.70 UA/mL). All human experimental procedures were reviewed and approved by the Ethics Committee on Clinical Research, Sakuragaoka Campus, Kagoshima University.
Caucasian blood samples were obtained from participants in the Benaroya Research Institute Registry and Repository. This study was approved by the Benaroya Research Institute Institutional Review Board (IRB) (Protocol IRB07109–431). The patients with allergies were age- and sex-matched with healthy controls. All PBMC samples were obtained under approved research protocols with informed consent. PBMCs were isolated by diluting the blood 1:1 in PBS and were prepared by density-gradient centrifugation according to standard protocols.
DC and skin cell preparations
For DC preparation, spleen and dLNs were collected, cut into small pieces, and digested in 10% FBS in RPMI containing 100mg/ml Liberase TL (Roche) and 100mg/ml DNase I (Roche) for 30min at 37°C and 200rpm in a shaking incubator. Skin cells were prepared from the ears using the gentleMACS^™^ Octo Dissociator (Miltenyi Biotec) in the RPMI solution containing 250 mg/ml Liberase TL (Roche) and 50mg/ml DNase I (Roche). Single cells were isolated using a cell strainer.
CAS mouse model and active cutaneous anaphylaxis (ACA)
For the CAS treatment, the bilateral ear skin was treated topically with MC903 (2mM in 100% ethanol) and OVA (100 μg, grade V; Sigma, St. Louis, MO) (100μg in 20 μL of PBS) daily for ten days to sensitize the mice. Unsensitized control mice were treated with vehicle (100% ethanol). For systemic sensitization, mice were given an i.p. injection of an NP16-OVA conjugate (100μg in PBS) and alum adjuvant (FUJIFILM Wako Pure Chemical Co., Tokyo, Japan) (9 μg) on day 26. For ACA analysis, mice were subcutaneously challenged with NP16-OVA on day 56, and rectal temperature was measured every 5 minutes to assess anaphylaxis.
Flow cytometry and immunohistological staining
Mouse and human cells were stained with the reagents for flow cytometry staining listed in S-Table 2. Flow cytometry was performed on a FACSCalibur and FACS Melody ^18^, and data were analyzed using FlowJo (Tree Star, CA, USA). For immunohistological staining, tissue sections were fixed with 4% PFA overnight at 4°C and then penetrated with a 10% sucrose solution. Fixed tissue was frozen in OCT compound (4583, Sakura Finetek, Japan), and 8-μm sections were prepared by cryostat (Leica, Wetzlar, Germany) and fixed onto slide grasses. After permeabilization (0.1% Triton X-100) and blocking (3% BSA-PBS), the sections were stained with the reagents for immunohistological staining listed in S-Table 2. Images were acquired using a fluorescence microscope BZ-X710 (Keyence, Osaka, Japan).
ELISA and antibody affinity assay
OVA- or NP-specific IgG1 and IgE concentrations were measured by ELISA using the following protocols. Plates were coated with OVA (100μg/ml), NP29-BSA (10μg/ml), or NP1-BSA (10μg/ml for IgG1, 0.5μg/ml for IgE) in 0.1M carbonate-bicarbonate buffer overnight at 4°C. After washing, 5% BSA in PBS was used to block nonspecific binding, and serum samples were added and incubated for 1 hour at 37°C. The following is the list of secondary antibodies: Anti-IgG1-HRP (A90–105p; BETHYL), anti-IgE-biotin (553419; BD), and StAv-HRP (43–4323; Invitrogen). BD OptEIA (555214, BD) was used for detection. Anti-OVA IgE was measured with a Mouse IgE ELISA OVA kit (DS Pharma Biomedical) according to the manufacturer’s protocol.^2^
Mass Cytometry by time of flight (CyTOF) analysis
Spleen, dLNs, skin cells, and PBMC were prepared from NS, CAS, and CAS+NP-OVA-treated mice. Cells (3×10^6^) were stained with 33 metal-conjugated antibodies (S-Table 3), Maxpar OnDemand Mouse Immune Profiling Panel Kit (Standard Biotools Cat:9200001) in a final volume of 100μl Maxper Cell Staining Buffer. Anti-IL-13R Ab was labeled with 154Sm by a Maxpar^®^ X8 Antibody Labeling Kit (201154A). Cells were incubated for 30 min at room temperature with 1uM Rhodium (Standard Biotools Cat:201103A) and 5 μl of FcR Block (Biolegend Cat:422301) to exclude dead cells and nonspecific binding of antibodies. Cells were washed twice with Maxper Cell Staining Buffer and were resuspended in 1ml of 1.6 % PFA (Thermo Fisher Scientific Cat: 28906) in PBS and then incubated at RT for 10min. Cells were acquired on a CyTOF mass cytometer to Helios specifications (CyTOF/Heslious) (Standard Biotools). 0.1× EQ Four Element Calibration Beads (Fluidigm) were added to the samples for data normalization of the FCS files using the CyTOF software. A total of 100,000 events were recorded for each sample and analyzed on Cytobank.
In the data analysis, positive events were first identified and gated. Then, non-viable cells and equalization beads were excluded to isolate live cells using FlowJo software (version 10.4.2). For clustering DC subsets and CD4 T cell subsets, CD3^−^ CD19^−^ CD11c^+^ MHC-II^+^ F4/80^−^ Ly6C^−^ cells and CD3^+^ CD19^−^ TCRb^+^ TCRgd^−^ CD4^+^ CD8^−^ cells were defined as DC and CD4 T cells respectively. We then down-sampled the live cells to 1000 cells per sample and down-sampled the DC and CD4 T cell populations to 500 cells each. These down-sampled files were then loaded into the R Catalyst package (version 1.22.0) and integrated with metadata. Subsequently, we performed FlowSOM (Flow Self-Organizing Maps) clustering using the following parameters: Features set to “type”, the mapping dimensions at x.dim=10 and y.dim=10, seed value “1234”, and the maximum number of clusters (max.k) set at 20 for live cells and 10 for DC and CD4 T cells. Following the FlowSOM clustering, we employed t-Distributed Stochastic Neighbor Embedding (tSNE) to visually represent our data, utilizing the runDR and plotDR functions.
Single-cell RNA-seq library preparation, sequencing, and data analysis.
According to a previous report, the data for the DC scRNAseq was generated by BD Rhapsody TAS-Seq.^59^ Briefly, cells were stained by BD sample tag (BD Biosciences), and CD11c^+^ MHCII^+^ Lin^−^ (CD3, B220, NK1.1, Ly-6G, TER119) splenic cells were sorted using a BD FACS Melody Cell Sorter. Then, 20,000 live cells were loaded onto the BD Rhapsody cartridge; cDNA trap and amplification were performed using the TAS-Seq method. Sequencing was performed with an Illumina Novaseq 6000 sequencer (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions (Read1: 56 base-pair, read 2: 155 base-pair). The pooled library concentration was adjusted to 1.75 nM, and 1% PhiX control library v3 (Illumina) was spiked into the library. Pair-end Fastq files (R1: cell barcode reads, R2: RNA reads) were processed as follows. Adapter trimming, quality filtering, and phase-shift base removal of sequencing data were performed by using Cutadapt 4.1(https://doi.org/10.14806/ej.17.1.200). Filtered cell barcode and cDNA reads were annotated and mapped to a reference genome (build GRCm39 release-107) by using STARsolo (2.7.10a)^60^ and known BD Rhapsody cell barcodes by the following parameters: –outSAMmultNmax 1–outFilterScoreMinOverLread 0 –outFilterMatchNminOverLread 0–outFilterMultimapScoreRange 0 –seedSearchStartLmax 30–soloType CB_UMI_Complex –soloUMIdedup NoDedup –soloMultiMappers Rescue –soloFeatures Gene GeneFull – soloCBmatchWLtype EditDist_2 –soloUMIlen 8 –soloCBposition 0_0_0_8 0_13_0_21 0_26_0_34 –soloUMIposition 0_35_0_42. Results from non-umi count data were converted to genes x cells matrix files, and the inflection threshold of the barcode rank plot was detected by using the DropletUtils package in R version 4.2.1.^61, 62^ Valid cell barcodes, and backgrounds from knee-point threshold to inflection threshold * 1.1 were separated by the Dropkick package (https://github.com/KenLauLab/dropkick).^63^ Associated sample tag reads were mapped to known barcode fasta by using bowtie2–2.4.2^64^ by the following parameters: -p 2 -D 20 -R 3 -N 0 -L 14 -i S,1,0.75 –norc –seed 656565 –reorder –trim-to 3:40 –score-min L,−9,0 –mp 3,3 –np 3 –rdg 3,3. Then, the cell barcode information of each read was added to the bowtie2-mapped BAM files and read counts of each tag in each cell barcode were counted using mawk. Results from the count data were converted to Tags x cells matrix file using the “data. Table” package in R version 4.2.1. For the assignment of each tag to each cell barcode, read counts of each tag in each valid cell barcode, which are defined by the cDNA matrix, were extracted from the tag/cell barcode expression matrix. Unassigned cell barcodes were labeled as “not-detected” cells. Then, a sum of the total read counts of each tag was normalized to 10M reads and the log2 fold-change between the first most counted tags and the second most counted tags within each cell barcode. Doublet cells (double-positive cells of any pair of Tags) were identified by the flowDensity package; (https://www.bioconductor.org/packages/release/bioc/html/flowDensity.html) and log2 fold-change between the first and second most counted tags under 0.30906 were identified as doublets. Finally, the remaining cell barcodes were assigned to the first most counted tags. Results from the tag-assigned count matrix file were processed through the R package Seurat 4.1.0.^65^ We first created a Seurat object for each dataset using the CreateSeuratObject function. We then filtered out mitochondrial gene-high cells (over 25%) and performed global normalization using the normalizeData function (scale.factor = 1,000,000). Next, highly variable genes of each dataset were identified using the FindVariableFeatures function (selection.method=mvp, mean.cutoff=c (0.1, Inf), dispersion.cutoff=c(0.5, Inf)). PCA analysis was performed based on the 8983 highly variable genes identified by the FindVariableFeatures function, and the data were scaled using the ScaleData function. Next, the enrichment of each PC was calculated using the JackStraw function, and statistically significant PCs (p≤1×10^−5^) were selected for clustering and dimensional reduction analyses. After performing linear dimensional reduction using PCA by the RunPCA function, the cells were clustered by the FindNeighbors function (dims=1:69) and the FindClusters function (resolution=0.6). Then, we performed linear dimensional reduction using Uniform Manifold Approximation and Projection (UMAP) by the RunUMAP function. Statistically significant marker genes between two groups were identified using the FindMarkers function (min.pct=0.25). Cell type estimation was performed using the SingleR package.^66^
For scRNAseq of T cells, PI^−^ Vb5.1^−^5.2^+^ CD4^+^ splenic OTII T cells were sorted into 96 well plates using a FACS Melody^18^ and then lysed in a buffer containing dNTPs and oligo(dT)-tailed oligonucleotides with a universal 5’-anchor sequence. A reverse transcription reaction, which adds 2–5 untemplated nucleotides to the cDNA 3’ end, was then performed. A template-switching oligo carrying 2 riboguanosines and a modified guanosine was added to produce an LNA as the last base at the 3’ end. After the first-strand reaction, the cDNA was amplified using 25 cycles. Next, Tn5 tagmentation was used to construct sequencing libraries from the amplified cDNA (Nextera XT DNA Library Preparation Kit, Illumina). The sequencing was carried out using NovaSeq6000 equipment (150PE, Illumina). Gene expression obtained in scRNAseq experiments was counted using the featurecounts (version 2.0.1) and an mRNA database based on the mouse RefSeq reference (mm 10). Then, we created a Seurat object from each gene expression matrix using CreateSeuratObject function. We normalized mRNA expression with log-normalize using normalizeData function (scale. factor = 10,000). Then, the top 2,000 most highly variable genes were identified by using FindVariableFeatures function. After scaling the data by ScaleData function, we performed linear dimensional reduction using PCA by RunPCA function. Then, the cells were clustered by FindNeighbors function (1:30) and FindClusters function (resolution=0.8). We performed linear dimensional reduction using UMAP. Statistically significant marker genes between two groups were identified using the FindMarkers function (min.pct=0.25).
Quantification and statistical analysis.
Distributed group data were expressed as mean ± standard deviation (SD) and statistical significance was determined using the two-tailed Student’s t-test. Data were analyzed with GraphPad Prism 7 software (GraphPad Software, La Jolla, CA, USA). All data in the present works were normally distributed. Differences were recognized as significant with a p-value of <0.05.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gowthaman U. Identification of a T follicular helper cell subset that drives anaphylactic Ig E. Science 365 (2019).10.1126/science.aaw 6433 PMC 690102931371561 · doi ↗ · pubmed ↗
- 2Harada Y. Type 2 helper T cells convert into Interleukin-13-expressing follicular helper T cells after antigen repriming. Translational and Regulatory Sciences 5, 1–12 (2023).
- 3Leyva-Castillo J.M., Hener P., Jiang H. & Li M. TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J Invest Dermatol 133, 154–163 (2013).22832486 10.1038/jid.2012.239 · doi ↗ · pubmed ↗
- 4Ohshima Y. Early sensitization to house dust mite is a major risk factor for subsequent development of bronchial asthma in Japanese infants with atopic dermatitis: results of a 4-year followup study. Ann Allergy Asthma Immunol 89, 265–270 (2002).12269646 10.1016/S 1081-1206(10)61953-9 · doi ↗ · pubmed ↗
- 5Nakajima H. & Takatsu K. Role of cytokines in allergic airway inflammation. Int Arch Allergy Immunol 142, 265–273 (2007).17124428 10.1159/000097357 · doi ↗ · pubmed ↗
- 6Holgate S.T. & Polosa R. Treatment strategies for allergy and asthma. Nat Rev Immunol 8, 218–230 (2008).18274559 10.1038/nri 2262 · doi ↗ · pubmed ↗
- 7Kubo M. Mast cells and basophils in allergic inflammation. Curr Opin Immunol 54, 74–79 (2018).29960953 10.1016/j.coi.2018.06.006 · doi ↗ · pubmed ↗
- 8Li M. Induction of thymic stromal lymphopoietin expression in keratinocytes is necessary for generating an atopic dermatitis upon application of the active vitamin D 3 analogue MC 903 on mouse skin. J Invest Dermatol 129, 498–502 (2009).18650845 10.1038/jid.2008.232 · doi ↗ · pubmed ↗