When Copper Meets Autoimmunity: A Rare Coexistence of Wilson's Disease and Systemic Lupus Erythematosus
Suresh Bhagoowani, Uooja Devi, Aqsa Munir, Ummulkiram Hasnain, Saad Khan, Javed Iqbal, Tehseen Akhtar

TL;DR
A rare case of Wilson's disease and lupus in a 17-year-old highlights the diagnostic challenges and severe complications from overlapping symptoms.
Contribution
This case report highlights the rare coexistence of Wilson's disease and systemic lupus erythematosus and emphasizes the need for early diagnosis.
Findings
The patient exhibited overlapping symptoms of Wilson's disease and systemic lupus erythematosus, leading to diagnostic challenges.
Despite treatment, the patient's condition worsened due to complications from a blood transfusion reaction.
Early recognition of Wilson's disease symptoms is critical to prevent severe outcomes in coexisting autoimmune conditions.
Abstract
The coexistence of systemic lupus erythematosus (SLE) and Wilson's disease (WD) is exceedingly rare, posing significant diagnostic challenges due to overlapping clinical presentations. Neurological and psychiatric manifestations commonly associated with WD can obscure or mimic symptoms of other systemic diseases, complicating accurate diagnosis and management. We report the case of a 17‐year‐old female diagnosed with WD and concurrent SLE, treated at Civil Hospital, Karachi, Pakistan. The patient initially presented with neuropsychiatric symptoms typical of WD, confirmed by diagnostic findings indicative of hepatic copper accumulation. She later developed significant proteinuria, hematuria, fever, and positive autoimmune serologies, raising suspicion of concurrent SLE. Despite therapeutic interventions, including methylprednisolone (1 mg), the patient's condition deteriorated, and she…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
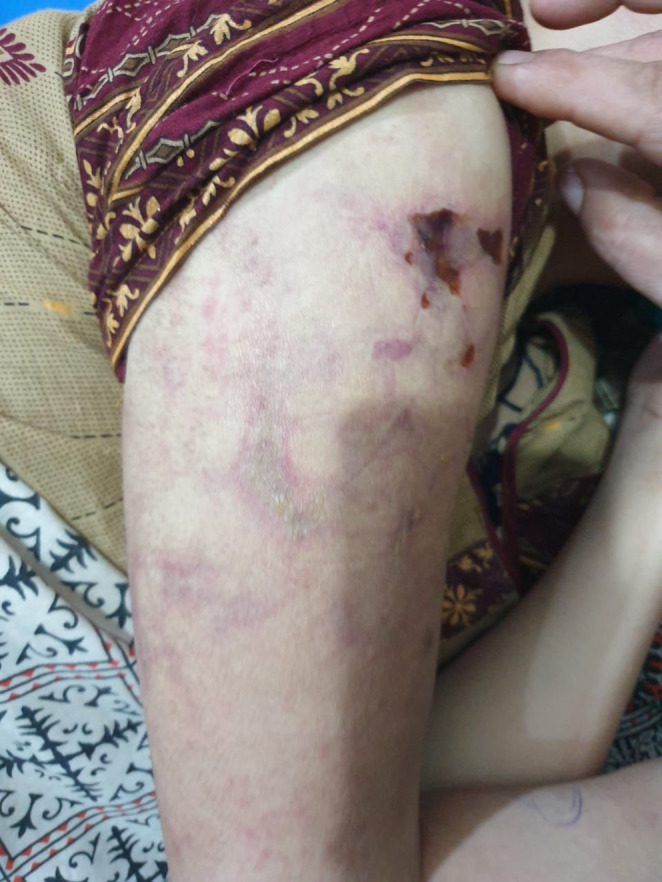 FIGURE 1
FIGURE 1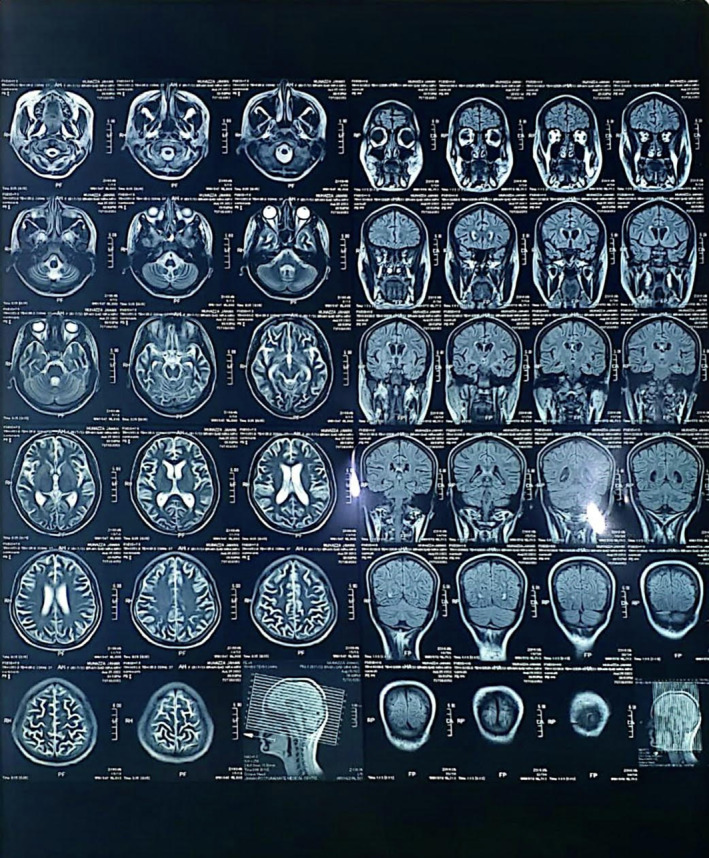 FIGURE 2
FIGURE 2| S.no | Study | Year | Age/Sex | Clinical features | Investigations | Management | Prognosis |
|---|---|---|---|---|---|---|---|
| 1. | Lishan Xu et al. [ | 2021 | 35‐year‐old female | Asymptomatic with known SLE, Routine follow‐up revealed concerning findings on liver ultrasound. |
Albumin: 33 g/L Other LFTs: Normal Ceruloplasmin: < 0.1 g/L Serum copper: 2.44 mol/L 24‐h urine copper levels: 110 g/24 h KF ring: Seen on slit‐lamp examination Low complement levels and positive antibodies for ANA and Anti‐dsDNA Abdominal ultrasound and CT: Suggestive of liver cirrhosis |
Zinc sulfate, methylprednisolone and hydroxychloroquine | US and labs showed marked improvement on the 11th‐month follow‐up. Ceruloplasmin levels raised to 0.20 g/L. |
| 2. | Yang et al. [ | 2024 | 9‐year‐old female | Hematuria |
LFTs: Deranged Albumin: 25.7 g/L Ceruloplasmin: < 9.5 mg/dL Low complement levels, ANA, and anti‐dsDNA: Positive Abdominal US: Normal Kidney Biopsy: Positive for antibodies Tumor markers: Negative Liver biopsy: Findings of fibrosis, and cirrhosis | Liver transplantation | The patient died 4 days post‐surgery |
| 3. | Zhang et al. [ | 2018 | 18‐year‐old female | Impaired speech and unusual limb movements for 2 years |
LFTs: Deranged Albumin: 32 g/L Ceruloplasmin: 0.033 g/L KF ring: Seen on slit‐lamp examination MRI Head: anomalous signals in the thalamus and basal ganglia. ANA, anti‐SSA antibody, anti‐rRNP antibody: raised CT abdomen: widespread hepatic lesions with splenic and liver enlargement |
IV Zinc sulfate and IV sodium dimercaptopropane sulfonate. Oral hydroxychloroquine, aspirin, and IV methylprednisolone. 1 month later, steroids decreased, and oral drugs replaced IV ones. |
At 1 month follow‐up. neurological symptoms and labs significantly improved Her symptoms did, however, return 6 months later |
| 4. | Hadef et al. [ | 2021 | 12‐year‐old male | Pale skin, yellow sclera and, Haematuria |
LFTs deranged Ceruloplasmin and copper levels: Below normal Urinary copper: Raised Kidney biopsy: lupus nephritis Anti‐DNA and anti‐PCNA: Positive | Steroids, cyclophosphamide, hydroxychloroquine, mycophenolate mofetil along with copper‐chelating therapy | Symptoms initially improved, but other neurological symptoms emerged after 2 years |
| Hb | 6.6 mg/dL |
| TLC | 2 × 109/L |
| Plt | 40/mcL |
| Serum Albumin | 2.2 g/dL |
| Globulin | 4.4 g/dL |
| LDH | 1027 U/L |
| Ferritin | 452 μg/L |
| Serum ceruloplasmin | 0.2 g/L |
| 24 h urinary copper | 204 μg/day |
| PT | 10.5 |
| INR | 1.18 |
| Total bilirubin | 11.5 mg/dL |
| SGOT | 1292 U/L |
| SGPT | 161 U/L |
| ALP | 261 U/L |
| GGT | 346 U/L |
| A/G ratio | 0.39 |
| BUN | 9 mg/dL |
| Cr | 0.7 mg/dL |
| Na | 136 mEq/L |
| K | 3.7 mEq/L |
| Cl | 107 mEq/L |
| Ca | 7.2 mEq/L |
| Clinical domains points | Clinical domains points |
|---|---|
|
|
Leukopenia |
| 3 points | |
|
Fever |
Thrombocytopenia |
| 2 points | 4 points |
|
|
Autoimmune hemolysis |
|
Nonscarring alopecia | 4 points |
| 2 point |
|
|
Oral ulcers |
Proteinuria > 0.5 g/24 h |
| 2 points | 4 points |
|
Oral ulcers |
Class 2 or 5 lupus nephritis |
| 2 points | 8 points |
|
Subacute cutaneous or discoid lupus |
Class 3 or 4 lupus nephritis |
| 2 points | 10 points |
|
Acute cutaneous lupus |
|
| 6 points |
Anticardiolipin IgG > 40 GPL or Lupus anticoagulant |
|
| 2 points |
|
Synovitis in at least 2 joints or tenderness in At least 2 joints and at least 30 min of Morning stiffness | |
| 6 points |
Low C3 or low C4 |
|
| 3 points |
|
Delirium |
Low C3 and low C4 |
| 2 points | 4 points |
|
Psychosis |
|
| 3 points |
Anti‐dsDNA antibodies |
|
Seizures | 6 points |
| 5 points |
Anti‐smith antibodies |
|
| 6 points |
|
Pleural or pericardial effusion | |
| 5 points | |
|
| |
|
Acute pericarditis | |
| 6 points |
| Clinical or laboratory findings | Points | |
|---|---|---|
| Kaiser fleischer rings | Present | 2 |
| Absent | 0 | |
| Neurological symptoms or MRI findings | Severe | 2 |
| Mild | 1 | |
| Absent | 0 | |
| Serum ceruloplasmin levels (g/L) | < 0.1 | 2 |
| 0.1–0.2 | 1 | |
| > 0.2 | 0 | |
| 24‐h urinary copper | > 2× upper limit of normal | 2 |
| 1–2× upper limit of normal | 1 | |
| Normal | 0 | |
| Normal, but > 5× upper limit of normal after D‐penicillamine | 2 | |
| Coombs‐negative hemolytic anemia | Present | 1 |
| Absent | 0 | |
| Total liver copper level (micromole/g) | > 5× upper limit of normal (> 4) | 2 |
| Increased (0.8–4) | 1 | |
| Normal (< 0.8) | −1 | |
| Rhodamine‐positive granules present | 1 | |
| Genetic mutation | Present on both chromosomes | 4 |
| Present on one chromosome | 1 | |
| Absent | 0 | |
| Total score | Diagnosis established | 4 |
| Diagnosis likely | 3 | |
| Diagnosis unlikely | 2 or less | |
- —Qatar National Library 10.13039/100019779
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTrace Elements in Health · Contact Dermatitis and Allergies · Aluminum toxicity and tolerance in plants and animals
Summary
- This case highlights the rare co‐occurrence of Wilson's disease and systemic lupus erythematosus (SLE), which complicates diagnosis due to overlapping symptoms.
- Treatment adjustments confirmed the SLE diagnosis, but the patient ultimately died from complications related to a blood transfusion reaction.
- The case emphasizes the need for careful diagnosis and management in complex systemic diseases.
Introduction
1
Wilson's disease (WD) is a genetic disorder caused by mutations in the ATP7B gene on chromosome 13, resulting in an autosomal recessive ailment. The poor excretion of copper by the biliary system is the primary anomaly associated with WD [1]. In Wilson's illness, there is a gradual rise in copper levels in the liver, eventually leading to an overflow, causing a multi‐organ involvement, including neurological and renal abnormalities. Dysfunction of ATP7B and hepatic dysfunction may also result in the buildup of other metals, like iron and manganese, within an individual's brain, possibly exacerbating symptoms of Wilson's illness [1]. Systemic lupus erythematosus (SLE) is an autoimmune condition that is more common in females and spares no organ. Organ damage manifests as immune system abnormalities and environmental, hormonal, and hereditary factors. The wide range of clinical symptoms includes manifestations in the skin and mucous membranes, musculoskeletal system, blood, heart and lungs, kidneys, and central nervous system. Lupus nephritis and neuropsychiatric lupus are regarded as the most severe types of organ involvement and can lead to a considerably shortened lifespan [2].
Seldom reports of SLE exacerbated by WD or the coexistence of two illnesses have been made. Here, we report a case of a young female patient suffering from both Wilson and SLE. A summary of Patients with WD and SLE from the Literature Review are presented in Table 1.
Case History and Examination
2
A 17‐year‐old girl with no known comorbid condition came to the Civil Hospital Karachi with complaints of fever on and off for 4 months, yellow discoloration of skin and sclera for 20 days, and odd behavior and irrelevant talk for the past week. The patient was alright 4 months back when she started to develop a fever, intermittent and undocumented, relieved by taking antipyretics; there is no specific time for fever; it was associated with rigor, chills, body ache, and joint pain but not accompanied by sore throat, burning micturition, cough, and loose stools. For the last 20 days, the patient has been complaining of yellow discoloration of the sclera that progressed to the whole body associated with dark‐colored urine, accompanied by a purplish rash all over the body not related to pale stool, itching, abdominal pain, melena, or hematemesis. A few days later, she developed odd behavior with irrelevant talk but remained conscious without any neurological symptoms. Past medical history was insignificant. She has taken multiple antipyretics prescribed by her local GP—no significant transfusion history. Personal history is positive for disturbed sleep and decreased appetite.
Investigation, Diagnosis, and Treatment
3
On initial examination, she looked sick and jaundiced, a young female average‐built conscious but disoriented with time, place, and person. She had a blood pressure of 100/60 mm of hg, pulse rate of 113 bpm, regular with a normal volume respiratory rate of 20 bpm. She had severe anemia, deep jaundice, mild dehydration, leukonychia, a puffy face with bilateral non‐tender parotid enlargement, oral ulcers, and angular stomatitis. Furthermore, on skin examination, the purplish rash looks like a reticular pattern with few necrotic plaques present on multiple sites (Figure 1). Except for the irrelevant talk, other neurological examinations were normal, and abdominal examination showed mild hepatosplenomegaly. The rest of the examinations were unremarkable. She is the only child of her consanguineous parents. The father was on antipsychotic medication for 20 years. Following admission, routine tests showed anemia (Hb = 6.6 mg/dL; normal = 12–16 mg/dL), leukopenia (TLC = 2; normal = 4–11 × 10^9^/L), thrombocytopenia (platelets = 40; normal = 150–450/mcL), elevated LDH (1027; normal = 140–280 U/L), decreased albumin (2.2; normal = 3.4–5.5 g/dL), raised globulin (4.4; normal = 2–3.5 g/dL), elevated total bilirubin (11.5; normal = 0.2–1.2 mg/dL), and SGOT (1292; normal = 8–45 units/L). Urine DR showed hematuria (8 − 10/hpf) and trace protein. Other lab findings, including thyroid profile, iron profile, HbA1C, and uric acid, were within normal ranges. Virologic tests were normal or negative; ANA titers were > 7.5 U/mL (normal = < 1.2 U/mL), anti‐dsDNA titers were 95.4 (normal = < 20 IU/L), RA factor > 20 IU/mL, and direct Coombs polyspecific 2+. C3 and C4 levels were low, and anti‐Ro/SSA antibodies were positive. Her serum ceruloplasmin was 0.2 (normal = 0.2–0.3 g/L), and 24‐h urine copper was 204 (normal < 60 micrograms/day). Ultrasound and Fibroscan were performed, revealing a normal ultrasound and a Fibroscan result of 3 kPa, with no indication of liver cirrhosis at diagnosis. Brain MRI showed a dilated ventricular system with minimal subependymal seepage (Figure 2), while MRA and MRV were unremarkable. EEG showed diffused 7–8 Hz alpha waves. Additional liver parameters, kidney function, and lab findings are detailed in Table 2.
Purplish Rash And Reticular Pattern With Necrotic Plaques.
MRI Of the Brain Revealing A Dilated Ventricular System With Minimal Subependymal Seepage.
Based on the laboratory findings of hemolytic anemia, lymphopenia, leukopenia, serositis, positive ANA, and anti‐dsDNA antibodies, the diagnosis of SLE was established according to the American College of Rheumatology revised criteria for the classification of SLE (Table 3). According to Leipzig criteria (Table 4), which include neurological symptoms, low serum ceruloplasmin, and increased 24‐h urinary copper, A score of 4 establishes a diagnosis of WD. Further findings suggested that the final diagnosis for this patient was acute hepatitis due to WD with secondary SLE. We initiated treatment with Slumedrol at a dose of 1 g for 5 days, alongside Rapricort at 1 mg/kg with a tapering schedule over 5 days. Additionally, Cap trientine 300 mg and zinc acetate 50 mg were administered twice daily. On day two, the patient developed seizures with a GCS drop to 11/15, leading to the initiation of Levetiracetam 500 mg twice daily for seizure control. Despite these efforts, the patient, unfortunately, did not survive, due to severe infection caused due to immunosuppressed state secondary to her autoimmune conditions, with trientine and zinc acetate continued until her passing.
Conclusions and Results
4
In conclusion, the co‐occurrence of WD and SLE presents a rare and complex diagnostic challenge due to overlapping symptoms, particularly neuropsychiatric and renal manifestations. This case highlights the importance of careful differential diagnosis and individualized treatment strategies when managing patients with multiple systemic conditions. Despite initial therapeutic improvement, the unfortunate outcome due to a blood transfusion reaction underscores the need for close monitoring of potential complications in such complex cases. Clinicians should maintain a high index of suspicion for autoimmune diseases in patients with WD, especially when they present with atypical symptoms.
Discussion
5
Mucormycosis is an aggressive fungal infection, often associated with significant morbidity and mortality. This case is unusual as the patient had no predisposing factors like diabetes or immunosuppression. Trauma is an infrequent trigger for mucormycosis, but it likely provided an entry point for the Rhizopus species in this case. This case demonstrates that early surgical intervention and antifungal therapy are crucial for favorable outcomes. This report reinforces the need for clinicians to consider mucormycosis even in immunocompetent pediatric patients presenting with unexplained ocular symptoms post‐trauma.
This case is exceptional in that there are no predisposing factors, highlighting the fact that even immunocompetent people can get this potentially fatal infection after a minor injury. A similar case was reported in another study in which a 31–year–old male underwent dental surgery, developed necrotizing lesions, and was later on diagnosed with mucormycosis [3]. The dental profession is responsible for being aware of the possibility of potentially severe and potentially fatal complications in healthy individuals [4]. The use of corticosteroids is also a risk factor for mucormycosis. The high death rate, even with improvements in diagnoses, emphasizes the necessity of greater awareness and prompt action in at‐risk groups [7]. In healthy hosts, neutrophils that destroy hyphal components by oxidative burst and macrophages that prevent spore germination are the defensive mechanisms. Infections in diabetics are caused by macrophage dysfunction [8]. Another study shows that GI mucormycosis can occur in malnourished and immunocompetent patients, so if malnourished patients exhibit abdominal symptoms and risk factors, clinicians should consider GI mucormycosis [9]. In this case, the symptoms of the patient were suggestive of rhabdomyosarcoma. The diagnosis was confirmed on histopathology. a study says that in immunocompetent patients' Nonspecific symptoms make diagnosis difficult; biopsy and culture are required [1]. In immunocompromised patients with liver cirrhosis, posaconazole can be used safely and effectively to treat mucormycosis [10].
Though rare, an association between WD and SLE has been reported, with the underlying mechanisms still not fully understood [10]. Antos et al. [11] note that SLE induced by D‐penicillamine can also manifest in patients undergoing treatment for WD. This case underscores the diagnostic complexity of WD, particularly when overlapping autoimmune conditions are present [5]. The patient's initial presentation with jaundice and anemia, followed by neurological symptoms, underscores the importance of considering WD in the differential diagnosis, particularly in the presence of liver dysfunction and autoimmune markers. Understanding the rare association between WD and SLE could provide insights into the pathophysiology of these conditions and improve management strategies [3].
Author Contributions
Suresh Bhagoowani: supervision, writing – original draft, writing – review and editing. Uooja Devi: writing – original draft, writing – review and editing. Aqsa Munir: writing – original draft, writing – review and editing. Ummulkiram Hasnain: writing – original draft, writing – review and editing. Saad Khan: writing – original draft, writing – review and editing. Javed Iqbal: writing – original draft, writing – review and editing. Tehseen Akhtar: supervision, writing – review and editing.
Ethics Statement
Ethical approval does not imply to case reports from institutional IRB.
Consent
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A. Członkowska , T. Litwin , P. Dusek , et al., “Wilson Disease,” Nature Reviews Disease Primers 4, no. 1 (2018): 21, 10.1038/s 41572-018-0018-3.PMC 641605130190489 · doi ↗ · pubmed ↗
- 2M. F. Shaikh , N. Jordan , and D. P. D'Cruz , “Systemic Lupus Erythematosus,” Clinical Medicine 17, no. 1 (2017): 78–83, 10.7861/clinmedicine.17-1-78.28148586 PMC 6297589 · doi ↗ · pubmed ↗
- 3Z. Yang , Y. Su , M. Liu , L. Sun , F. Zhang , and W. Lin , “A Rare Concomitance of Wilson's Disease and Systemic Lupus Erythematosus in a Teenage Girl: A Case Report and Literature Review,” Frontiers in Pediatrics 11 (2024): 1296426, 10.3389/fped.2023.1296426.38259603 PMC 10800781 · doi ↗ · pubmed ↗
- 4L. Xu , B. Liu , Z. Liu , et al., “Co‐Occurrence of Wilson's Disease and Systemic Lupus Erythematosus: A Case Report and Literature Review,” Biomed Research International 21, no. 1 (2021): 229, 10.1186/s 12876-021-01814-5.PMC 813902434020599 · doi ↗ · pubmed ↗
- 5Y. Zhang , D. Wang , W. Wei , and X. Zeng , “Wilson's Disease Combined With Systemic Lupus Erythematosus: A Case Report and Literature Review,” Bio Med Research International 18, no. 1 (2018): 85, 10.1186/s 12883-018-1085-5.PMC 600298229907136 · doi ↗ · pubmed ↗
- 6D. Hadef , S. Slimani , F. Lahouel , B. Benlahcen , and N. Bouchair , “Concomitance of Primary Systemic Lupus Erythematosus and Wilson's Disease in a Male Child: A Case Report,” Rheumatology 61, no. Supplement_2 (2022): 079, 10.1093/rheumatology/keac 496.079. · doi ↗
- 7Z. Yang , Q. Li , S. Liu , Z. Zong , L. Yu , and S. Sun , “Systemic Lupus Erythematosus Combined With Wilson's Disease: A Case Report and Literature Review,” Bio Med Research International 24, no. 1 (2024): 253, 10.1186/s 12887-024-04713-2.PMC 1101753038622515 · doi ↗ · pubmed ↗
- 8O. Bandmann , K. H. Weiss , and S. G. Kaler , “Wilson's Disease and Other Neurological Copper Disorders,” Lancet Neurology 14, no. 1 (2015): 103–113, 10.1016/S 1474-4422(14)70190-5.25496901 PMC 4336199 · doi ↗ · pubmed ↗