Dynamic kinetic resolution-mediated synthesis of C-3 hydroxylated arginine derivatives
Ye Zheng, Zhenyu Chen, Guy J. Clarkson, Stephen A. Marshall, Jianliang Xiao, Christopher J. Schofield, Martin Wills, Andrew V. Stachulski

TL;DR
Scientists developed a new method to synthesize hydroxylated arginine derivatives using a dynamic kinetic resolution process.
Contribution
A novel dynamic kinetic resolution method is introduced for synthesizing C-3 hydroxylated arginine derivatives.
Findings
The DKR method produced a (3R)-hydroxylated product matching a synthetically derived compound.
High diastereomeric and enantiomeric excess was achieved using an (R)-SEGPHOS/Ru(II) catalyst.
The approach demonstrates DKR's effectiveness for amino acid derivative synthesis.
Abstract
Hydroxylated amino acids and their derivatives, including those found in proteins, are important in biology and medicinal chemistry. Incubation of N-acetyl-l-arginine with clavaminic acid synthase, a key oxygenase in clavulanic acid biosynthesis, affords a (3R)-hydroxylated product that is identical to material obtained by total synthesis from Boc-beta alanine. The key step employed dynamic kinetic resolution (DKR) of a β-ketoester precursor, achieved in high diastereomeric and enantiomeric excess using an (R)-SEGPHOS/Ru(II) catalyst. The results highlight the utility of DKR for the preparation of C-3 hydroxylated amino acid derivatives.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
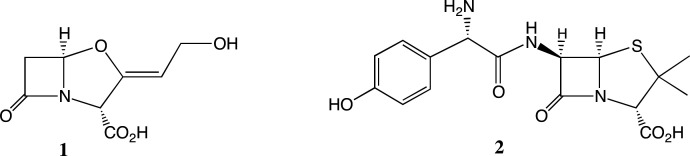 Figure 1
Figure 1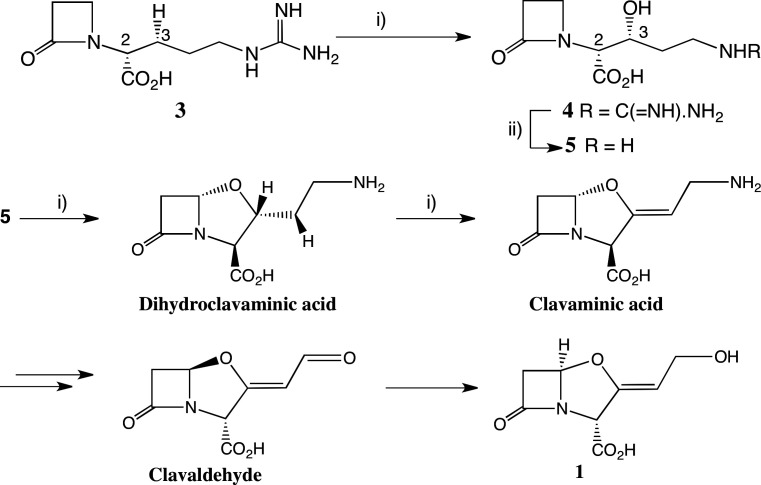 Figure 2
Figure 2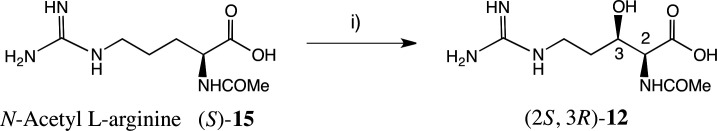 Figure 3
Figure 3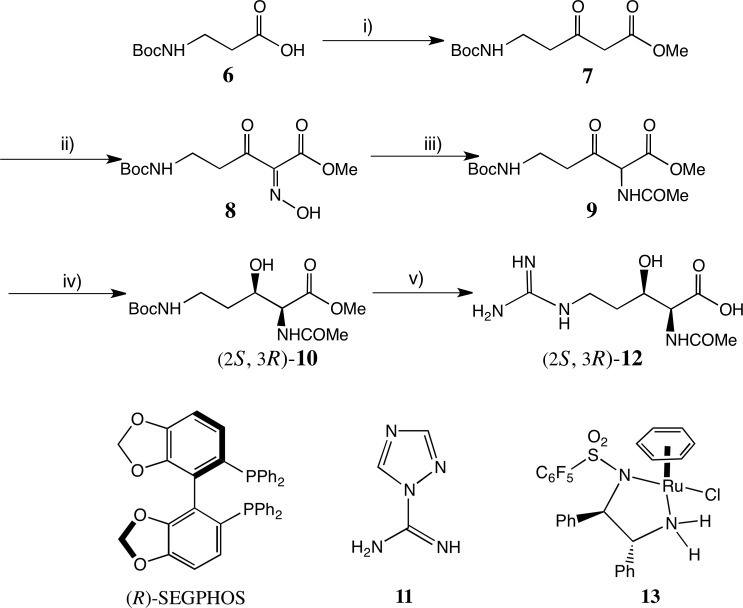 Figure 4
Figure 4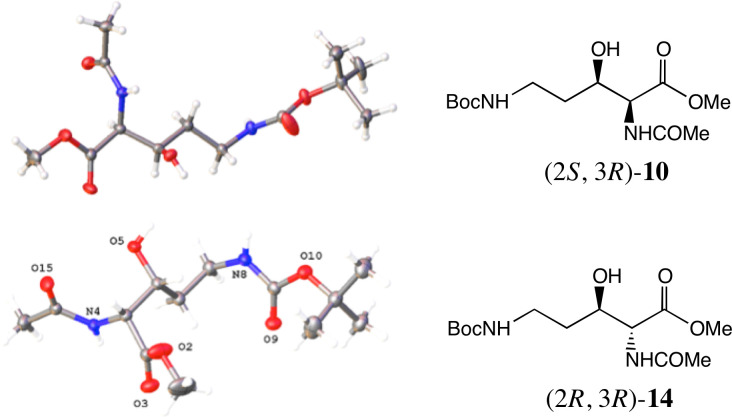 Figure 5
Figure 5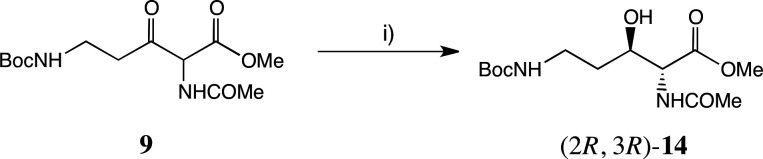 Figure 6
Figure 6- —CCT
- —Johnson Mattheyhttp://dx.doi.org/10.13039/501100023964
- —European Regional Development Fund (ERDF)
- —Science City Project
- —Wellcome Trusthttp://dx.doi.org/10.13039/100010269
- —Biotechnology and Biological Sciences Research Councilhttp://dx.doi.org/10.13039/501100000268
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAsymmetric Hydrogenation and Catalysis · Synthetic Organic Chemistry Methods · Chemical Synthesis and Analysis
Introduction
The discovery of the Streptomyces metabolite clavulanic acid 1 (CA, figure 1) [1–3] was an important advance in antibacterial chemotherapy. Although 1 is a weak antibacterial per se, it is a potent inhibitor [4] of a range of nucleophilic serine beta-lactamases produced by bacteria as a defence against many beta-lactam antibiotics [4,5]. The combination of amoxicillin 2 with 1, as augmentin [6] and its generic formulations, is still a vital broad-spectrum, orally absorbed antibacterial treatment. The growing threat of antimicrobial resistance, in particular via bacterial strains producing beta-lactamases [7,8], has led to a renaissance of interest in 1 and newer beta-lactamase inhibitors [9,10].
Clavulanic acid 1 and amoxicillin 2, the constituents of augmentin.
There is no effective synthesis of 1, which is commercially obtained by fermentation. An early published synthesis of rac**-1** afforded a yield of less than 5% in a key step [11–14]. Given its importance, there has been surprisingly little structure-activity study of derivatives of CA 1, especially of late. The biosynthesis of 1, however, is known in some detail [14]: three early steps are catalysed by the oxygenase clavaminic acid synthase (CAS) (figure 2). A C3 hydroxyl group, the eventual source of the ring oxygen of 1, is stereoselectively introduced into deoxyguanidino-proclavaminic acid 3 by CAS catalysis [15,16]. The guanidino group of the resultant product 4 is then hydrolysed by an amidino hydrolase (PAH) [17,18] to afford proclavaminic acid 5, a substrate for two further CAS-catalysed reactions, that is oxidative ring closure followed by desaturation. Eventually, the final CAS product clavaminic acid is converted in further biocatalysed steps, which are not fully elucidated, to clavaldehyde, which on reduction delivers CA 1 [14].
Important steps in clavulanic acid biosynthesis. (i) Clavaminate acid synthase (CAS.Fe(II)); each CAS catalysed reaction is coupled to conversion of O2, 2-oxoglutarate to CO2, succinate; (ii) proclavaminate amidino hydrolase (PAH.Mn(II)), H2O; urea coproduct.
CAS is a member of the 2-oxoglutarate (2OG)-dependent superfamily of oxygenases [19], members of which catalyse a remarkably wide range of oxidative reactions, including Lys- and Arg-residue C−3 hydroxylations. For example, Lys C−3 hydroxylation is catalysed by JMJD7 [20,21] and Arg C−3 hydroxylation is catalysed by JMJD5 [22], both of which are potential cancer drug targets [23].
There is a need for efficient syntheses of C-3 hydroxylated amino acids, in order to validate structures, including stereochemistry, of biosynthetic/natural products, to act as starting points for chemo-enzymatic syntheses of drugs such as CA 1, and to use in the generation of antigens to create antibodies selective for post-translational modifications introduced by 2OG oxygenases. In addition to the CA 1 biosynthetic intermediates, CAS also catalyses the hydroxylation of N-acetyl Arg to give N-acetyl-(2S,3R)-3-hydroxy-Arg (see figure 3), further exemplifying the biocatalytic potential of 2OG oxygenases, first demonstrated with proline hydroxylases [24–26]. However, only a single stereoisomer is produced in the CA catalysed reaction, so that its use in preparing stereoisomers is limited. Here, we report the use of dynamic kinetic resolution (DKR) for the stereoselective synthesis of both (2S,3R)- and (2R,3R)-N-acetylated C-3 hydroxy-Arg derivatives.
Biohydroxylation of N-acetyl-l-arginine (s)−15. Conditions: (i) Clavaminate acid synthase (CAS.Fe(II)); the reaction is coupled to conversion of O2, 2OG to CO2, succinate.
We considered that the core α-amino, β-hydroxy units of 4 and 5 could be accessible as single enantiomers via DKR [27–29] of an appropriate β-keto precursor. Indeed, the homologous 3-hydroxylysine derivatives have been prepared in this manner [30,31]. Similarly, we have shown that MeBmt, sc. (2S,3R,4R,6E)-3-hydroxy−4-methyl−2-(methylamino)−6-octenoic acid, the unusual hydroxy amino acid of cyclosporin A, can be efficiently obtained by asymmetric transfer hydrogenation (ATH) of a β-ketoanilide bearing an N-Me substituent, giving the desired anti-product [32]. Other syntheses of 5 involved a lengthy chiral pool approach [33] or a non-stereospecific aldol reaction [34].
The relative syn-(2S,3R)-stereochemistry in 4 and 5 could result from addition to a keto precursor under the classical Noyori hydrogenation conditions [27], namely an appropriate Ru catalyst and a high pressure of H_2_. The transfer hydrogenation mode developed by Somfai [35] and others was predicted to give anti-products, namely (2S, 3S)/(2R, 3R); we studied both modes. Work on a related ketoester substrate studied by Ishida, Touge et al. [36,37] indicated that hydrogenation using [NH_2_Me_2_][(RuCl((S)-Segphos®))2(µ-Cl)3] delivered the syn product, while an oxo-tethered derivative (DENEB) of a Noyori–Ikariya catalyst [(arene)Ru(TsDPEN)Cl] gave the anti-product.
The keto precursor for all our DKR studies was efficiently obtained, as shown in figure 4 (full details are in the electronic supplementary material). Two-carbon homologation of Boc-β-alanine 6 using Masamune’s conditions using a malonate half ester, as adapted by Genet [31], gave a very good yield of β-ketoester 7 [38]. Standard nitrosation catalysed by AcOH [31] afforded the crystalline oxime 8 again in very good yield. Following the hydrogenation of the oxime, the intermediate amine was acylated with PhCOCl, affording a crystalline product, but in rather low yield. Instead, by using Ac_2_O as both solvent and reagent, the acetamide 9 resulted in an excellent yield. This proved a useful substrate for all the hydrogenations performed.
Syntheses of (2S,3R)-3-hydroxy N-acetyl Orn and Arg derivatives. Reagents: (i) CDI, KO2CCH2CO2Me, MgCl2, THF, 76%; (ii) NaNO2, AcOH, 75%; (iii) H2/Pd, Ac2O, 95%; (iv) [NH2Me2][(RuCl((R)-Segphos®))2(µ-Cl)3], CH2Cl2-MeOH, 72%; (v) (a) LiOH, aq. MeOH. (b) CF3CO2H, (c) 11, aq. Na2CO3, purification via HPLC.
Asymmetric hydrogenation (AH) of 9 using (R) or (S)-[NH_2_Me_2_][(RuCl(Segphos®))2(µ-Cl)3] [39,40] proved to be the method of choice for the formation of chiral syn products. Thus, hydrogenation of 9 at 50 atm H_2_ with (R)-[NH_2_Me_2_][(RuCl(Segphos®))2(µ-Cl)3] afforded (2S,3R)−10 in 72% yield (99.6% ee, d.r. 89:11 in favour of the 2,3-syn product) and this was reproduced on a 1 g scale. This is a good d. r. considering the lack of branching of the C(4) methylene of 9 [35]. Recrystallization from EtOAc-hexane afforded (2S,3R)−10 as a single stereoisomer whose absolute configuration was confirmed by X-ray crystallography (figure 5) [41].
Single crystal X-ray structures of (2S,3R)-methyl 2-acetamido−5-((tert-butoxycarbonyl)amino)−3-hydroxypentanoate 10 and (2R,3R)-methyl 2-acetamido-5-((tert-butoxycarbonyl)amino)-3-hydroxypentanoate 14.
To introduce the guanidino substituent present in the enzymic hydroxylation product (figure 2), the Me ester was cleaved using LiOH to minimize the risk of α-epimerization. The Boc group was cleaved from the resulting acid with CF_3_CO_2_H: the product was taken up in aq. Na_2_CO_3_ solution and treated with amidino triazole reagent 11 [42] to afford arginine derivative 12 [24–26]. This reagent appears superior to the earlier amidinopyrazole [43] and allows steady reaction at 20°C.
Other hydrogenation conditions were also explored (see electronic supplementary material for full details, including chiral HPLC analyses). AH of 9 under identical conditions to the above, but using the (S)-SEGPHOS-based catalyst, generated (2R,3S)- or ent−10 in 75% yield, 99.8% ee, d.r. 95:5. ATH of 9 was achieved using formic acid as the hydrogen donor, and a range of Noyori–Ikariya catalyst derivatives (see electronic supplementary material for full details) [44–49]. The most selective catalyst was the variant 13 [50] with a C_6_F_5_ group on the DPEN unit (figure 4). With the catalyst containing the (R,R)-ligand, the main product was (2R,3R)-14 (72% yield, 99% ee, d.r. 9.7:1), namely, the *anti-*diastereoisomer of 10 above (figure 6). Following recrystallization, a single stereoisomer was produced, whose structure was also confirmed by single crystal X-ray crystallography (figure 5).
ATH of 9 using catalyst (R,R)-13 to form (2R,3R)-14. Reagents: (i) 1 mol% 15, HCO2H/Et3N (5:2), DCM, 72 h, rt, 72%.
The method was readily scaled up to afford gram quantities of the desired product (2S,3R)-10. Following conversion to the arginine derivative (2S,3R)-12, the material was shown by ^1^H NMR to be identical to the product of incubation of commercial N-acetyl-l-arginine 15 with CAS (figure 3, see electronic supplementary material for full details). It was known [25,26] that N-acetyl-l-arginine 15 is a superior CAS substrate to the corresponding Orn compound as it affords clean hydroxylation without the formation of an alkene byproduct.
In summary, DKR is an excellent tool for the synthesis of key C-3 hydroxylated Arg derivatives and can readily deliver gram quantities suitable for both structural/stereochemical assignment studies and for incorporation into peptides for use in antibody generation. There is clear scope for variation of the Nα-acyl substituent, which may eventually enable a chemoenzymatic synthesis of CA 1 and its derivatives.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cole M, Howarth TT, Reading C. 1976 Beta-lactam antibiotic from streptomyces clavuligerus, DE 2517316, to Beecham, 1975. Chem Abs 84, 72635 t.
- 2Brown AG, Butterworth D, Cole M, Hanscomb G, Hood JD, Reading C, Rolinson GN. 1976 Naturally-occurring .BETA.-lactamase inhibitors with antibacterial activity. J. Antibiot. 29, 668–669. (10.7164/antibiotics.29.668)950324 · doi ↗ · pubmed ↗
- 3Howarth TT, Brown AG, King TJ. 1976 Clavulanic acid, a novel β-lactam antibiotic isolated from Streptomyces clavuligerus. JCS Chem Commun 266.
- 4Reading C, Cole M. 1977 Clavulanic Acid: a Beta-Lactamase-Inhibiting Beta-Lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 11, 852–857. (10.1128/aac.11.5.852)879738 PMC 352086 · doi ↗ · pubmed ↗
- 5Spratt BG, Jobanputra V, Zimmermann W. 1977 Binding of Thienamycin and Clavulanic Acid to the Penicillin-Binding Proteins of Escherichia coli K-12. Antimicrob. Agents Chemother. 12, 406–409. (10.1128/aac.12.3.406)334066 PMC 429926 · doi ↗ · pubmed ↗
- 6Ball AP, Geddes AM, Davey PG, Farrell ID, Brookes GR. 1980 Clavulanic acid and amoxycillin: a clinical, bacteriological, and pharmacological study. Lancet 315, 620–623. (10.1016/s 0140-6736(80)91118-6)6102627 · doi ↗ · pubmed ↗
- 7Paterson DL, Bonomo RA. 2005 Extended-Spectrum β-Lactamases: a Clinical Update. Clin. Microbiol. Rev. 18, 657–686. (10.1128/cmr.18.4.657-686.2005)16223952 PMC 1265908 · doi ↗ · pubmed ↗
- 8Brem J et al. 2016 Structural basis of metallo-β-lactamase, serine β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun 7, 12406.27499424 10.1038/ncomms 12406 PMC 4979060 · doi ↗ · pubmed ↗