Whole-genome sequencing of the Streptomyces coelicolor bldA39 mutant (J1700) reveals hundreds of previously unknown mutations
Jack W. Stone, John T. Munnoch, Paul A. Hoskisson

TL;DR
This paper reveals the genome of a historically significant Streptomyces coelicolor mutant, uncovering hundreds of new mutations.
Contribution
The study identifies over 300 previously unknown mutations in a key Streptomyces strain used for decades in research.
Findings
The bldA39 mutation (T>C) was confirmed in the S. coelicolor J1700 genome.
Over 300 additional mutations were discovered, including those in biosynthetic gene clusters.
The study clarifies the genetic background of the J1501 strain used to create J1700.
Abstract
We report the genome sequence of the bldA39 (J1700) mutant of Streptomyces coelicolor, a historically important strain that is deficient in sporulation and antimicrobial production. The S. coelicolor J1700 strain was used extensively from the 1980s onwards to underpin important discoveries in development and antibiotic production in Streptomyces. The bldA gene encodes a leucyl tRNA, required for the translation of the rare TTA codon found in ~2% of genes in Streptomyces. The whole genome of S. coelicolor J1700 was obtained via Illumina sequencing and mapped to the S. coelicolor M145 reference genome. Analysis of the genome sequence compared to S. coelicolor M145 identified the known bldA39 mutation (T>C) and revealed more than 300 further mutations, likely associated with the S. coelicolor J1501 genetic background the strain was created in, including the nature of the hisA1 and uraA1…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
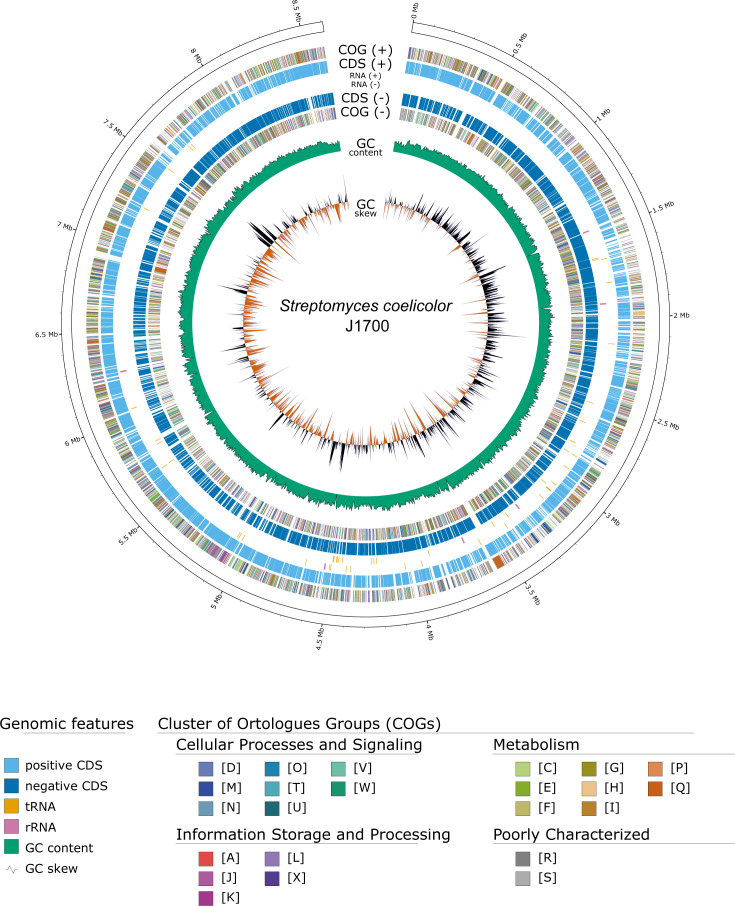 Fig. 1
Fig. 1 Fig. 2
Fig. 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Natural Products and Biosynthesis · Genomics and Phylogenetic Studies · RNA and protein synthesis mechanisms
Data Summary
This whole-genome sequencing project has been deposited in NCBI under the Bioproject PRJNA1186139. The WGS reads used can be accessed in the NCBI’s SRA under the accession number SAMN44744323. Table S1 is available on Figshare 10.6084/m9.figshare.27798405[1].
Introduction
The bacterial genus Streptomyces has long been studied as a model for morphological differentiation and the production of natural products such as antibiotics [2]. Decades of genetic analysis of Streptomyces bacteria have enabled the identification of regulatory mechanisms that are essential for morphological development (formation of unigenomic spores on reproductive structures called aerial hyphae) and antibiotic production [35]. During these studies, numerous mutants have been isolated that are blocked at distinct stages of development, and these fall into two main classes: the so-called white (whi) mutants, which are able to form aerial mycelium but are unable to complete development into mature spores. The second class are the so-called bald (bld) mutants, which are blocked at an earlier stage of development, which prevents the erection of the aerial hyphae and subsequent development of spores. In addition to causing the loss of aerial mycelium, several mutations in bld loci have been found to pleiotropically block antibiotic production [67].
Amongst the most severe bld phenotypes that have been identified to date were associated with the bldA locus, where mutations result in complete loss of morphological development and natural product production [89]. The bldA locus was the first morphological mutant mapped by Hopwood [10] as bldA1 (S48), with further mapping efforts of Merrick [11] characterizing 12 bld mutants into 4 mapping groups, 5 of which were bldA alleles. The bldA locus was cloned by Piret and Chater [12] and subsequently shown to encode a leucyl tRNA, required for the translation of the rare TTA codon found in ~2% of genes in Streptomyces [1314]. The Leu-tRNA^UUA^ accumulates late in growth [131516], with much of the bldA-associated phenotype believed to be mediated via the highly conserved, TTA-codon containing global transcriptional regulator, AdpA [717]. The effect that bldA disruption has on the control of antibiotic production has also been attributed to TTA codons present in biosynthetic gene cluster (BGC) situated regulators such as actII-ORF4, redD (in Streptomyces coelicolor [1819]) and ccaR (in Streptomyces clavuligerus [20]).
Amongst the original bldA mutants characterized by Merrick [11] was the bldA39, a mutation that was subsequently used in phage cloning experiments to transfer the mutation [12] to the S. coelicolor J1501 strain background (his1A, ura1A, strA1, pgl-1, SCP1^−^, SCP2^−^ [21]) that was historically used for genetic mapping experiments. This created the S. coelicolor J1700 (bldA39) strain that was subsequently used in studies by Leskiw et al. [1416] to characterize the bldA gene. The genetic lesion leading to the bldA morphological phenotype can be complemented through the addition of a copy of the bldA gene on a phage [12] and by integrating plasmids (Stone, Munnoch and Hoskisson, unpublished). Studies of antibiotic production in the S. coelicolor J1700 (bldA39) strain found that there is reduced expression of genes in the undecylprodigiosin (red) BGC [21]. Actinorhodin (act) production appears to be predominantly regulated at the level of transcription, although translation fusions of the 5′ end of actII-ORF4 containing a single UUA codon to an ermE gene demonstrated that the bldA tRNA is present and functional early in growth [22].
Many of the studies to date on bldA have been conducted in the S. coelicolor J1700 (bldA39) strain; however, the wider genetic background of this strain is currently unknown. Here, we describe the genome sequencing of the S. coelicolor J1700 mutant and provide further information on additional mutations in that strain background. These data are deposited in NCBI under the Bioproject PRJNA1186139. The WGS reads (paired-end Illumina data) used can be accessed in the NCBI’s SRA under the accession number SAMN44744323.
Methods
S. coelicolor J1700 was grown for 24 h in Tryptone Soy Broth (TSB) media at 30 °C, shaking at 200 r.p.m. The genomic DNA of the strain was extracted according to Kieser et al. [23], and modifications were provided in Actinobase [24]. Sequencing was performed by Novogene using the Illumina NovaSeq 6000 platform. DNA sequence analysis enabled the mapping of the reads to the S. coelicolor M145 chromosome [25]. Breseq [26] mapping analysis of each strain was performed (using default settings, without predict-polymorphisms) and the output GenomeDiff files were compared (gdtools COMPARE). The analysis reports ‘predicted mutations’, including small variants (indels and single nucleotide changes), regions of ‘unassigned missing coverage evidence’ (typically large deletions) and ‘unassigned new junction evidence’ where multiple forms of the same sequence are suggested by the data (typically deletions with read coverage of the S. coelicolor M145 reference sequence also present). Mutations were then transferred to the reference genome (using gdtools APPLY) generating a FASTA, GENBANK and GFF3 version of the genome. This was carried out for all mutations in ‘predicted mutations’ while necessary manual edits were made as required. Auxotroph analysis was carried out according to Kieser et al. [23].
Results & discussion
S. coelicolor J1700 has extensive mutations across the genome that likely reflect the genotype of the parental strain J1501
The whole-genome sequence of S. coelicolor J1700 was determined at 137.6× coverage and was mapped to the wild-type S. coelicolor M145 strain (NC_003888.3) [22] (Fig. 1). The S. coelicolor J1700 strain was originally constructed in the S. coelicolor J1501 genetic background (his1A, ura1A, strA1, pgl-1, SCP1^−^, SCP2^−^) that was historically used for genetic mapping experiments [23]. The S. coelicolor J1700 strain was created through phage-mediated transfer of the bldA39 mutation [12] to S. coelicolor J1501, although the overall genetic background of the strain remains unknown.
GenoVi visualization of the S. coelicolor J1700 genome [35]. Labelling from outside to the inside: COGs [36] (forward strand); CDS, tRNAs and rRNAs (forward strand); CDS, tRNAs and rRNAs (reverse strand); COGs (reverse strand); Genome G+C content; Genome GC skew.
The genome of S. coelicolor J1700 was found to be 8 608 660 bp (Fig. 1), consisting of 7823 CDSs (compared to the 7846 CDSs in S. coelicolor M145 [25]). Following the Breseq analysis, an ‘unassigned missing coverage evidence’ region of the S. coelicolor J1700 genome was identified, which indicates a 53 414 bp deletion between 7 014 046 bp and 7 071 460 bp of the genome. This deletion results in the loss of the SCO6353-SCO6406 genes. Comparison with S. coelicolor M145 reveals that there are 324 mutations in J1700 (Fig. 1 and Table S1: 10.6084/m9.figshare.27798405, available in the online Supplementary Material). The mutations in S. coelicolor J1700 are characterized as 121 non-synonymous mutations, 78 synonymous mutations, 74 intergenic mutations, 39 coding frameshifts, 3 pseudogenes (SCO0634, SCO2890 and SCO4318), 3 deletions of ~1 kb (affecting SCO3991–SCO3991, [SCO4697]–[SCO4699] and SCO5630–[SCO5632], where the square brackets indicate a potential polar affect on that gene), 3 non-sense, 2 non-coding (including bldA and methionine tRNA anticodon CAT) and 2 non-stop mutations. It is likely that many of these mutations reflect those in the S. coelicolor J1501 genetic background in which J1700 was constructed [12].
The bldA39 mutation in S. coelicolor J1700 results in an anticodon change from Leu-UAA to Ser-UGA
Lawlor et al. [13] first showed that the bldA39 mutation results in a mutation in the anticodon loop of the Leucyl-tRNA^UUA^, which generates a putative seryl-anticodon. It is currently unclear if this tRNA species can be charged with serine by the cognate aminoacyl-tRNA synthetase (aaRS). Given the selectivity of aaRSs enzymes, this is unlikely as there are limited editing mechanisms in the aaRSs between the cognate tRNAs for serine and leucine [27]. The bldA39 mutant represents the only ‘classical’ bldA mutant strain that disrupted the tRNA anticodon, with other mutations affecting the anticodon stem of the tRNA*^bldA^* (bldA1 [nt 28 G-A]) and the tRNA*^bldA^* d-arm (bldA16 [nt 22 C-T], bldA62 [nt 23 A-C]) [21]. The single nucleotide T-C mutation attributed to the bldA39 phenotype is found at position 3 380 959 in S. coelicolor J1700 chromosome (position 3 380 943 in S. coelicolor M145).
Discrepancies in undecylprodigiosin expression on S. coelicolor J1700 may be the result of IS110 located in the BGC (red)
AntiSMASH [28] of the S. coelicolor J1700 genome revealed the presence of all 24 BGCs known from S. coelicolor M145. A detailed investigation of the BGCs indicated that there are several mutations within these gene clusters.
Guthrie and Chater [21] reported reduced red gene expression in the S. coelicolor J1700 strain using xylE transcriptional reporter strains. Examination of the S. coelicolor J1700 reveals the presence of a synonymous mutation in the undecylprodigiosin BGC pathway-specific regulator redD [19] (SCO5877: CTC-CTT; L150L). This reflects a change to a much less frequently used codon, but which is unlikely to impact significantly on red gene expression. More likely to affect transcription of the red cluster in S. coelicolor J1700 is the presence of an IS110 element [29] in the intergenic region between SCO5885 (putative membrane protein) and SCO5886 (redR, which encodes a 3-oxoacyl-[acyl-carrier protein] synthase II) at position 6 442 702 bp in the genome.
Further mutations in BGCs were noted, such as in the coelichelin BGC, with a synonymous mutation in a putative peptide synthetase (SCO0492; TTC-TTT; F2247F). Two non-synonymous mutations were noted in the calcium-dependent antibiotic (CDA) BGC in the CDA peptide synthetase I (SCO3230; CTC-GTC; L3479A; and GCC-GTC; A5927V). A non-synonymous mutation was also identified in the actinorhodin (act) BGC, in the ActIV bifunctional cyclase [second ring] thioesterase [30] (SCO5091; GCG-GAC; A689D). Additional mutations are also present in the coelimycin BGC [31], where two synonymous mutations are present in cpkPβ (SCO6269: GCG-GCC; A179A; and GCG-GCA; A166A) and two further synonymous mutations in cpkC (SCO6273: AAG-AAA; K562K; and GGG-GGC; G561G).
The consequences of these mutations are unknown; however, with mostly synonymous mutations present in the BGC genes, there are unlikely to be significant effects on the phenotype of S. coelicolor J1700, with extensive complementation studies required to assess potential effects on phenotype and through mRNA stability where synonymous changes are present.
The hisa1 genotype is a result of mutation in the histidinol dehydrogenase gene, hisD
One of the genetic markers present in S. coelicolor J1501 strain, the progenitor of the bldA39 strain J1700, is hisA1. Strains carrying this mutation are histidine auxotrophs [23]. The designation of hisA1 as a mapping group is well established, but the literature is not clear about where the mutation that results in histidine auxotrophy is situated. This may reflect the use of ‘hisA’ a complementation group in older work on S. coelicolor genetics. Work from Limauro et al. [32] suggests that the so-called hisA gene in S. coelicolor was in fact an ortholog of hisD, the histidinol dehydrogenase in Escherichia coli. Histidinol dehydrogenase catalyses the terminal reaction in histidine biosynthesis that oxidizes l-histindol to l-histidine and in S. coelicolor is the first gene in a three-gene operon (hisDCB). Sequencing of S. coelicolor J1700 identified a missense mutation in the gene hisD (T-C) resulting in an E264G change in histidinol dehydrogenase. This mutation maps to the region of the protein that coordinates a catalytic zinc ion that is required for substrate binding [33]. To confirm the requirement of S. coelicolor J1700 for histidine, growth on minimal media was tested for its ability to support S. coelicolor J1700 in the presence and absence of histidine, confirming auxotrophy (Fig. 2).
Auxotrophic analysis of the S. coelicolor J1700 strain. The S. coelicolor wild-type (M145) and bldA39 (J1700) strain were grown in the presence of histidine (his) or uracil (ura) according to Kieser et al. [23] to test for auxotrophy based on the genotype of the parental strain S. coelicolor J1501.
The uraA1 mutation maps to the putative uridine 5′-monophosphate synthase in S. coelicolor J1700
A further historic and widely used genetic marker in S. coelicolor J1501 is uraA1, where strains exhibit uracil auxotrophy. Analysis of the mutations detected in S. coelicolor J1700 revealed that there was no mutation present in the uraA gene, suggesting that this may also reflect the use of uraA as a complementation group designation rather than a gene designation. Auxotrophy analysis of the strain revealed S. coelicolor J1700 is auxotrophic for uracil (Fig. 2). Analysis of the genome mutations in S. coelicolor J1700 identified a putative uridine 5′-monophosphate synthase (SCO3650: pyrE) that possesses a frameshift mutation resulting in a 10 bp deletion (99–108/549 nt) towards the 5′ end of the CDS. The pyrE gene also maps to the uraA1 location of the physical map of the S. coelicolor chromosome [34], suggesting that it is this mutation that results in uracil auxotrophy in S. coelicolor J1700. Uridine 5′-monophosphate synthases catalyse the formation of uridine monophosphate as an initial step in uridine triphosphate biosynthesis and RNA metabolism. This led us to conclude that uracil auxotrophy is likely the result of a frameshift in pyrE of S. coelicolor.
Summary
The whole-genome sequencing of bacterial strains has revolutionized the way microbiology is conducted. The sequencing of historical strains that have underpinned significant discoveries in particular fields can help to shed light on discrepancies in older literature, such as here around discrepancies in red gene expression in S. coelicolor J1700, that could be attributed to the presence of a previously discovered insertion element. Furthermore, the clarification of genetic markers that were historically used to map mutations can help clarify the literature for researchers who may never have undertaken genetic mapping experiments. Overall, the sequencing of the historically important S. coelicolor J1700 bldA39 strain will provide a resource for researchers to use in studying development and antibiotic production in Streptomyces.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Stone J Munnoch J Hoskisson PA Whole-genome sequencing of the streptomyces coelicolor blda 39 mutant (J 1700) of reveals hundreds of previously unknown mutations. figshare Figshare 202410.6084/m 9.figshare.27798405.v 1 · doi ↗
- 2Schlimpert S Elliot MA The best of both worlds—Streptomyces coelicolor and Streptomyces venezuelae as model species for studying antibiotic production and bacterial multicellular development J Bacteriol 2023205 e 001532310.1128/jb.00153-2337347176 PMC 10367585 · doi ↗ · pubmed ↗
- 3Chater KF Taking a genetic scalpel to the Streptomyces colony Microbiology 19981441465147810.1099/00221287-144-6-146533789395 · doi ↗ · pubmed ↗
- 4Bush MJ Tschowri N Schlimpert S Flärdh K Buttner MJ c-di-GMP signalling and the regulation of developmental transitions in streptomycetes Nat Rev Microbiol 20151374976010.1038/nrmicro 354626499894 · doi ↗ · pubmed ↗
- 5Flärdh K Buttner MJ Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium Nat Rev Microbiol 20097364910.1038/nrmicro 196819079351 · doi ↗ · pubmed ↗
- 6Chandra G Chater KF Developmental biology of Streptomyces from the perspective of 100 actinobacterial genome sequences FEMS Microbiol Rev 20143834537910.1111/1574-6976.1204724164321 PMC 4255298 · doi ↗ · pubmed ↗
- 7Chandra G Chater KF Evolutionary flux of potentially bld A-dependent Streptomyces genes containing the rare leucine codon TTA Antonie van Leeuwenhoek 20089411112610.1007/s 10482-008-9231-518320344 · doi ↗ · pubmed ↗
- 8Chater KF Chandra G The evolution of development in Streptomyces analysed by genome comparisons FEMS Microbiol Rev 20063065167210.1111/j.1574-6976.2006.00033.x 16911038 · doi ↗ · pubmed ↗