High-Accuracy Long-Read Sequencing of Mycobacterium tuberculosis PSNK363 Isolated From the Democratic People's Republic of Korea
Thi-Binh Dang, Nackmoon Sung, Kyunghyun Lim, Soyoung Lee, Jaehyun Jeon, Sanghoon Jheon

TL;DR
This paper reports the first high-accuracy genome sequence of a Mycobacterium tuberculosis clinical strain from North Korea, revealing significant genetic differences compared to the commonly used H37Rv reference.
Contribution
The study provides the first high-fidelity genome assembly of M. tuberculosis PSNK363 from North Korea using long-read sequencing.
Findings
The PSNK363 genome is 10,578 bp longer than H37Rv and contains a large inversion region with deletions and insertions.
The PSNK363 genome has more protein-coding genes and higher potential for virulence and drug resistance analysis.
Abstract
Long-read sequencing is a valuable technique for high-precision genome analysis. Despite the widespread use of the Mycobacterium tuberculosis H37Rv genome sequence as a reference for genetic variation analysis, its suitability for comparing clinical strains is limited. Therefore, we constructed the first known whole genome of a clinical M. tuberculosis strain, PSNK363, isolated from the Democratic People's Republic of Korea, using high-quality high-fidelity (HiFi) read sequencing and compared its genetic variations to those of H37Rv. PSNK363 was cultured to obtain genomic DNA, which was subjected to de novo whole-genome assembly using PacBio Sequel II with long-read HiFi sequencing. The sequences were compared to the reference genome H37Rv. HiFi long-read sequencing of M. tuberculosis PSNK363, with an accuracy of 99.99%, revealed a single circular chromosome of 4,422,110 bp, which is…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1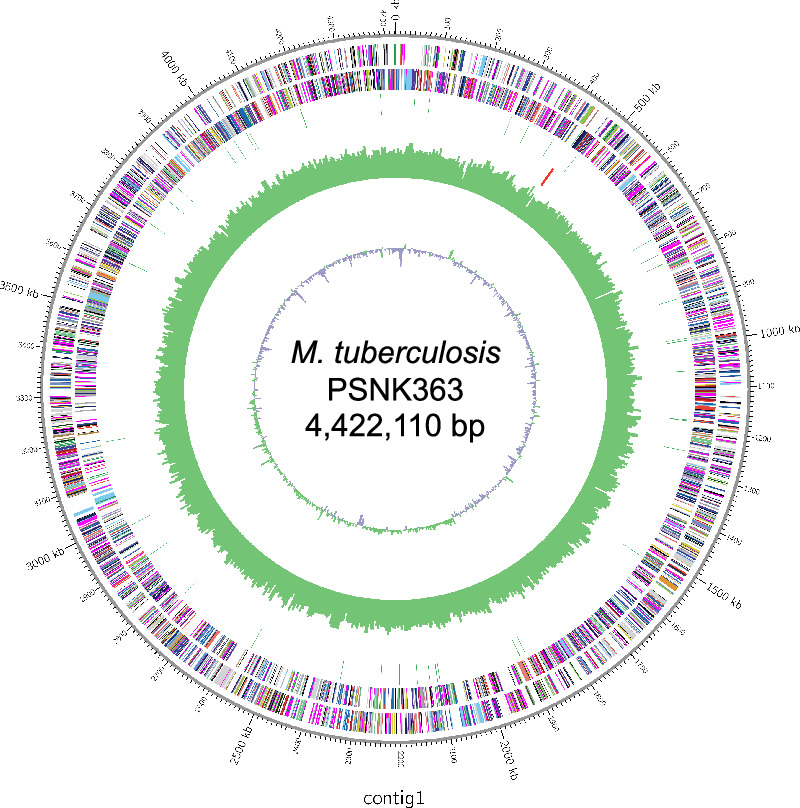 Figure 2
Figure 2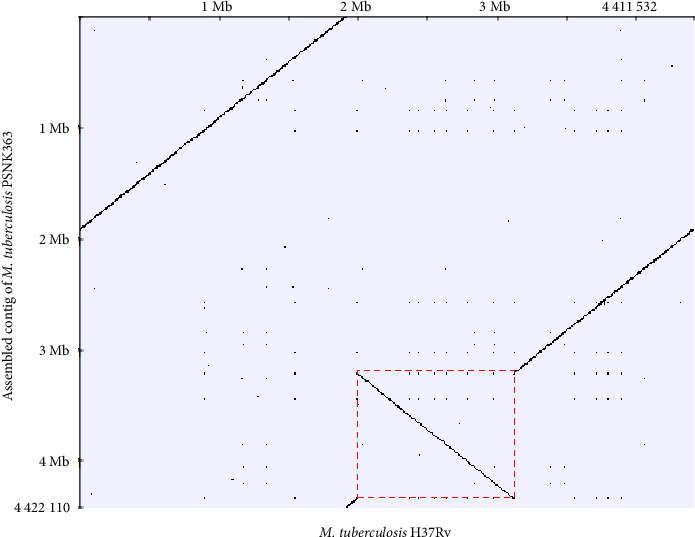 Figure 3
Figure 3- —Seoul National University Bundang Hospital
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMycobacterium research and diagnosis · Tuberculosis Research and Epidemiology · RNA and protein synthesis mechanisms
1. Introduction
Tuberculosis (TB) is a serious public health problem in the Democratic People's Republic of Korea (DPRK), with an estimated incidence rate of 513 per 100,000 people for TB and 5200 cases of multidrug-resistant (MDR)-TB in 2022 [1]. This places the country among the 30 high-burden countries for TB and MDR-TB/rifampicin (RIF)-resistant (RR)-TB [1]. Despite the availability of diagnostic tests and drugs for MDR-TB treatment, the regimens are more expensive and arduous than those for drug-susceptible TB, resulting in high treatment failure and mortality rates. A study from sanatoria in the DPRK [2] indicated a considerable percentage of drug-resistant TB isolates, while other studies [3, 4] raised concerns regarding the need to scale up MDR-TB treatment in the DPRK. While favorable treatment outcomes in patients with MDR-TB have been observed in the DPRK [3], the high number of TB and MDR-TB cases owing to challenges in accessing appropriate treatment medications [4] poses a risk of further spread. Additionally, the factors driving the MDR-TB epidemic in the DPRK remain unclear, and data on Mycobacterium tuberculosis genome sequencing in the DPRK are insufficient and unpublished.
To address this genomic data gap on M. tuberculosis in the DPRK, we performed whole-genome sequencing of a clinical sample obtained from the Eugene Bell Foundation collection [2]. The foundation transferred sputum samples of patients in the DPRK to Masan National TB Hospital (Masan, South Korea) from 2007 to 2009 for repetitive sequence-based polymerase chain reaction (rep-PCR) genotyping using the DiversiLab® system (bioMerieux). Subsequently, the strains were stored and provided to Seoul National University Bundang Hospital (SNUBH) for genomic sequencing. Among 179 M. tuberculosis isolates studied in this collection, only 7 (3.9%) were confirmed to be susceptible to drugs using the conventional method with Löwenstein–Jensen (LJ) medium. M. tuberculosis PSNK363 was derived from these susceptible isolates and belongs to the most prevalent cluster identified through rep-PCR.
Long-read sequencing was recognized as the “Method of the Year 2022” [5] owing to its exceptional potential in genome, transcriptome, and epigenome analyses. Specifically, the PacBio long-read sequencing is known for its high accuracy, rendering it suitable for applications that require precise single nucleotide variant (SNV) detection [6], such as genetic analysis of M. tuberculosis. Hence, in the present study, we employed highly accurate long-read high-fidelity (HiFi) sequencing to construct a whole-genome sequence of the unique M. tuberculosis strain PSNK363 from the DPRK and performed a comparative analysis with H37Rv.
2. Materials and Methods
2.1. Selection of M. tuberculosis PSNK363
M. tuberculosis PSNK363 was obtained from a collection of 179 clinical M. tuberculosis isolates maintained as part of our previous study [2], stored at Masan National TB Hospital (South Korea), and transferred to SNUBH, South Korea, for whole-genome sequencing.
2.2. Ethics Approval
This study was performed in line with the guidelines of SNUBH and approved by the Institutional Review Board (approval number: X-2008-631–901) and Institutional Ethics Committee (approval number: IBC-2002-A-001-02) of SNUBH.
2.3. M. tuberculosis Culture, Drug Susceptibility Testing, and DNA Preparation
M. tuberculosis PSNK363 was aerobically cultured in Middlebrook 7H9 medium containing 0.05% (v/v) Tween 80% and 10% Middlebrook OADC growth supplement (Sigma-Aldrich, St. Louis, MO, USA) at 37°C.
The sensitivity of PSNK363 to RIF (40 μg/mL), isoniazid (0.2 μg/mL), ethambutol (2.0 μg/mL), levofloxacin (2.0 μg/mL), ethionamide (40 μg/mL), amikacin (30 μg/mL), and kanamycin (40 μg/mL) was confirmed via conventional methods using LJ medium. Pyrazinamide sensitivity was determined using pyrazinamidase assay with the Wayne method.
Genomic DNA was isolated from M. tuberculosis using the cetyltrimethylammonium bromide method. Briefly, the bacilli in suspension were heat-treated at 80°C for 30 min in a water bath. After centrifugation, the cell pellets were resuspended in 500 μL of Tris-EDTA buffer (0.01 M Tris–HCl and 0.001 M EDTA [pH 8.0]). Subsequently, the cells were treated with lysozyme (50 mg/mL) for 1 h at 37°C and then with 10% sodium dodecyl sulfate and proteinase K (10 mg/mL) for 20 min at 60°C. Thereafter, 200 μL of N-acetyl-N, N,N,-trimethyl ammonium bromide was added to approximately 500 μL of the lysed cell suspension. The mixture was briefly vortexed and incubated for 10 min at 60°C. An equal volume of chloroform-isoamyl alcohol (24:1, v/v) was added to the mixture, which was then centrifuged for 5 min; 0.6 volume of isopropanol was added to the supernatant to precipitate the DNA. After cooling for 30 min at 20°C, the DNA solution was centrifuged for 15 min, and the pellet was washed once with 70% ethanol. Finally, the air-dried pellet was redissolved in 50 μL of 0.1× Tris-EDTA buffer and stored at −20°C until further use.
2.4. DNA Quality Control
PSNK363 gDNA was quantified using the Qubit® fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) with Broad Range dsDNA assay kits, and DNA purity was measured using the NanoDrop™ 2000/2000c spectrophotometer (Thermo Fisher Scientific). Fragment size was evaluated through agarose gel electrophoresis. PSNK363 gDNA adhered to the following criteria for sequencing: A260/280 ratio: 1.7–2.0; A260/230 ratio: 1.8–2.0; and DNA weight: > 5 μg/μL. The purified DNA was stored at 4°C prior to the preparation of sequencing libraries.
2.5. Whole-Genome Sequencing
The M. tuberculosis PSNK363 genome was sequenced using PacBio Sequel II (Pacific Biosciences, Menlo Park, CA, USA) at Macrogen Inc. (Seoul, Republic of Korea) with a long-read HiFi library (Figure 1). The genomic DNA was fragmented using dsDNA fragmentase (New England Biolabs, Ipswich, MA, USA) to achieve the desired size (10–25 kb) for library construction. The resulting DNA fragments were then processed using the SMRTbell Express Template Prep Kit 2.02 (Illumina Inc., San Diego, CA, USA), following the manufacturer's instructions.
2.6. Bioinformatics and Data Analysis
2.6.1. Genome Assembly
The generated PacBio sequencing reads (6,133,251 reads; total read length: 359,444,804,032 bp) and HiFi sequencing reads (1,621,177 reads; total read length: 23,913,685,936 bp) were assembled using the Phred/Phrap/Consed package and CLC Genomics Workbench v6.5 (CLC bio, Aarhus, Denmark). The resulting contigs from PacBio sequencing were scaffolded by sequencing the reads from the fosmid clones, and the gaps in the scaffolds were closed using PCR and Sanger sequencing. The contigs and Sanger sequence reads for gap closure were combined via manual curation using Phred/Phrap/Consed and CodonCode Aligner 3.7.1 (CodonCode Corp., Centerville, UT, USA). Genome assembly quality was assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO 5.1.3) [7, 8]. A graphical circular representation of the genome was visualized using Circos v0.69-9 [9].
2.6.2. SNVs and Small Insertion and Deletion (Indel) Analysis
HiFi reads were aligned to the reference H37Rv genome using minimap2 [10] to generate a Binary Alignment Map file, and Paftools.js was then applied to call the variants. Variant annotations were performed using SnpEff. The evolutionary genealogy of genes was functionally annotated using nonsupervised orthologous group (EggNOG) DB tools [11]. To generate the assembly used for gene prediction and annotation, Prokka 1.14.6 [12] was used with the genome assembly application. The predicted genes were functionally annotated using EggNOG v4.5, InterProScan 5.30–69.0 [13], and PSI-BLAST 2.10.0 [14].
2.6.3. Structural Variant Analysis
The alignment results from the assembled contigs of PSNK363 and H37Rv were aligned using nucmer lastz. Subsequently, these alignments were processed using Structural Variants from MUMmer to identify structural variants, including deletions, insertions, and inversions.
3. Results
3.1. Genome Features of M. tuberculosis PSNK363 and Its Comparison With H37Rv
M. tuberculosis PSNK363 harbors a single circular chromosome of 4,422,110 bp (Figure 2). It possesses an average G + C content of 65.6% and comprises 4079 protein-coding sequences, 53 tRNA genes, and three rRNA genes (Table 1). The total length of HiFi reads is 1,621,177 bp, N50 contig length with HiFi reads is 15,079 bp, and the mean read length is 14,750 bp; thus, the HiFi accuracy was 99.99%. The genome of PSNK363 is 10,578 bp longer than that of H37Rv (4,411,532 bp).
3.2. Functional Classification of Orthologous Clusters
In this study, the annotation of protein sequences predicted by Prokka was refined using EggNOG. Overall, the annotation revealed the presence of 4146 genes, among which 4079 corresponded to coding sequences. Among the proteins coded by these coding sequences, 4006 were successfully matched to entries in the EggNOG database. Notably, 3946 proteins were associated with a single EggNOG category, whereas 60 proteins were linked to multiple EggNOG categories. However, 73 proteins revealed no significant similarity to known proteins in the EggNOG database.
Table 2 presents the EggNOG category distribution of functional annotation results and their frequencies. More than 27% (1112 proteins) of the proteins were categorized as having unknown functions. Additionally, a smaller proportion of proteins (3.5%; 146 proteins) was assigned to the EggNOG category related to translation, ribosomal structure, and biogenesis.
3.3. SNV Indels and Variant Calling
To identify SNVs and small indels, SNV calling was performed by comparing our assembly with the H37Rv genome. The assembled contig of PSNK363 and H37Rv revealed 2180 variants comprising 1582 SNVs, 321 insertions, and 277 deletions.
To interpret the effect of SNVs, the World Health Organization (WHO) catalog of mutations in M. tuberculosis [16] was used as a reference to search for variants appearing in the PSNK363 genome. As presented in Table 3, among the 16 identified variants, 12 sequences were described in the WHO catalog, and four sequences were not previously reported. Sequence variants that matched those in the WHO database [16] belonged to Group 4 and were not associated with resistance-interim, whereas Group 5 was not associated with resistance. Variants in PSNK363, including eis_c-3520A > C, fgd1_c.651C > G, mmpL5_c-2915A > G, and Rv2752c_c.489A > G, are not listed in the WHO catalog.
3.4. Structural Variants
For large variants (> 50 kb), the PSNK363 assembly contained 28 structural variants: 10 insertions, 15 deletions, and 3 inversions (Table 4).
Upon comparison of the H37Rv and PSNK363 assemblies, an inversion sequence was observed in the syntenic dot plot (Figure 3). The inversion region of PSNK363 contains approximately 1,119,943 nucleotides at positions 3,217,011–4,336,953. The inversion sequence of PSNK363 results in two breakpoint regions in the genome sequence: a large breakpoint (Gap 1) at positions 4,336,953–4,345,124 and a smaller breakpoint (Gap 2) at positions 3,217,005–3,217,011. The inversion breakpoints were confirmed using PCR and resequenced using Sanger sequencing to assemble the PSNK363 genome (data not shown), which indicated the presence of an inversion in the PSNK363 chromosome.
4. Discussion
Whole-genome sequencing has revolutionized our understanding of drug resistance evolution, pathogenesis, and virulence in clinically relevant pathogens, such as M. tuberculosis. Single-molecule sequencing technology, particularly long-read sequencing, enables the generation of complete and closed microbial genome assemblies [17, 18]. This emerging technology can improve the accuracy of reference genome sequences and enable direct comparisons among different bacterial genomes without initially performing reference-based assembly [18, 19]. Although data on M. tuberculosis genomes from the DPRK are scarce [20], M. tuberculosis PSNK36 is the first strain with complete whole-genome isolated from patients with TB in a sanatorium in the DPRK using long-read sequencing.
Characterization of protein functions in M. tuberculosis is crucial for determining its pathogenicity, antibiotic resistance, and virulence. Our findings revealed that the functional annotation of PSNK363 is primarily associated with transcription (K); lipid transport and metabolism (I); replication, recombination, and repair (L); amino acid transport and metabolism (E); and energy production and conversion (C). These findings align with those of previous studies, which demonstrated a high percentage of genes involved in these groups across various M. tuberculosis populations [21, 22]. These findings suggest that these genes are typically conserved to ensure M. tuberculosis interactions with its host, particularly during mycobacterial persistence when host pathogens compete for nutrients and evade immune recognition [21]. Additionally, we classified SNVs related to resistance-conferring mutations of PSNK363 using the WHO mutation catalog [16]. We identified unique variants in PSNK363, including eis (c-3520A > C), fgd1 (c.651C > G), mmpL5 (c-2915A > G), and Rv2752c (c.489A > G), which were associated with susceptibility to certain genomic variants via a phenotypic method.
Gene inversion is a common phenomenon in bacterial evolution and can promote increased virulence and drug resistance in various pathogens [23, 24]. Large inversion events have been observed in some isolates belonging to the Beijing family [25–27] and KZN strains [28]. Our analysis revealed an inversion in the assembly of PSNK363, and notably, most indel events occurred around the inversion region. This finding suggests that inversions generate adaptive mutations at their breakpoints, resulting in increased frequency owing to the selection of breakpoint variants [29]. Although deletions [30, 31] and large inversions [25, 26] in M. tuberculosis have been reported, knowledge on the relationship between them remains limited. Given that high-accuracy, HiFi long-read sequencing effectively detects structural variants [32], we successfully assembled the inversion sequence and observed that this rearranged region harbored most deletion and insertion variants. Further investigations are needed to determine the effect of inversion events on genome evolution. Nevertheless, our study provides data that are not yet represented in the WHO catalog and primary data from whole-genome sequencing of M. tuberculosis in the DPRK.
5. Conclusions
In conclusion, our findings provide comprehensive insights into the genomic features of M. tuberculosis PSNK363, along with a description of its complete genome sequence and annotation. The genome of M. tuberculosis PSNK363 is approximately 4.4 Mbp with 4079 annotated genes, exhibiting a large-size inversion region. High-accuracy whole-genome sequencing of PSNK363 holds potential for enriching virulence databases and identifying informative loci for drug resistance analysis in M. tuberculosis isolates in the DPRK and other countries.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization Global Tuberculosis Report 2023 2023 World Health Organization
- 2Jung J. Jegal Y. Ki M. Characteristics of Drug Resistant Tuberculosis in Sanatoria of North Korea Journal of Korean Medical Science 20173271105111010.3346/jkms.2017.32.7.11052-s 2.0-8502018400928581266 PMC 5461313 · doi ↗ · pubmed ↗
- 3Seung K. J. Franke M. Linton S. W. Multidrug-resistant Tuberculosis Treatment in North Korea: Is Scale-Up Possible? P Lo S Medicine 201613810.1371/journal.pmed.10020622-s 2.0-84988916895 PMC 497071727483430 · doi ↗ · pubmed ↗
- 4Seung K. J. Linton S. W. The Growing Problem of Multidrug-Resistant Tuberculosis in North Korea P Lo S Medicine 201310710.1371/journal.pmed.10014862-s 2.0-84881246785 PMC 372792123935457 · doi ↗ · pubmed ↗
- 5Anonymous Method of the Year 2022: Long-Read Sequencing Nature Methods 202320 p. 110.1038/s 41592-022-01759-x 36635552 · doi ↗ · pubmed ↗
- 6Oehler J. B. Wright H. Stark Z. Mallett A. J. Schmitz U. The Application of Long-Read Sequencing in Clinical Settings Human Genomics 2023171 p. 7310.1186/s 40246-023-00522-3PMC 1041087037553611 · doi ↗ · pubmed ↗
- 7Seppey M. Manni M. Busco Z. E. M. Kollmar M. Assessing Genome Assembly and Annotation Completeness Gene Prediction: Methods and Protocols 2019 New York Springer 22724510.1007/978-1-4939-9173-0_142-s 2.0-8506532510731020564 · doi ↗ · pubmed ↗
- 8Waterhouse R. M. Seppey M. Simão F. A. BUSCO Applications From Quality Assessments to Gene Prediction and Phylogenomics Molecular Biology and Evolution 201835354354810.1093/molbev/msx 3192-s 2.0-8504557762229220515 PMC 5850278 · doi ↗ · pubmed ↗