Nonclinical and clinical characterization of the absorption, metabolism, and excretion of islatravir
Kerry L. Fillgrove, Randolph P. Matthews, Bing Lu, Yuexia Liang, Munjal Patel, Wen Liu, Catherine Z. Matthews, Yang Liu, S. Aubrey Stoch, Rosa I. Sanchez, Marian Iwamoto

TL;DR
Islatravir, a new HIV-1 treatment, is well absorbed and processed in the body, with promising properties for long-term use.
Contribution
The paper provides a comprehensive nonclinical and clinical characterization of islatravir's absorption, metabolism, and excretion.
Findings
Islatravir is well absorbed in both nonclinical species and humans after oral administration.
The elimination of islatravir involves oxidative deamination and renal excretion of the unchanged drug.
The active triphosphate form of islatravir is the most abundant intracellular species.
Abstract
The development of new and improved antiretroviral therapies that allow for alternative dosing schedules is needed for people living with HIV-1. Islatravir is a deoxyadenosine analog in development for the treatment of HIV-1 that suppresses HIV-1 replication via multiple mechanisms of action, including reverse transcriptase translocation inhibition and delayed chain termination. Islatravir is differentiated from other HIV-1 antiretrovirals by its high potency, long t½, broad tissue distribution, and favorable drug resistance profile. A comprehensive evaluation was performed to provide data on the mass balance, absorption, metabolism, and excretion of islatravir through studies in nonclinical species, and in adults without HIV-1 infection, using radiolabeled islatravir. Islatravir was well absorbed in both nonclinical species and humans following oral administration. The elimination of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
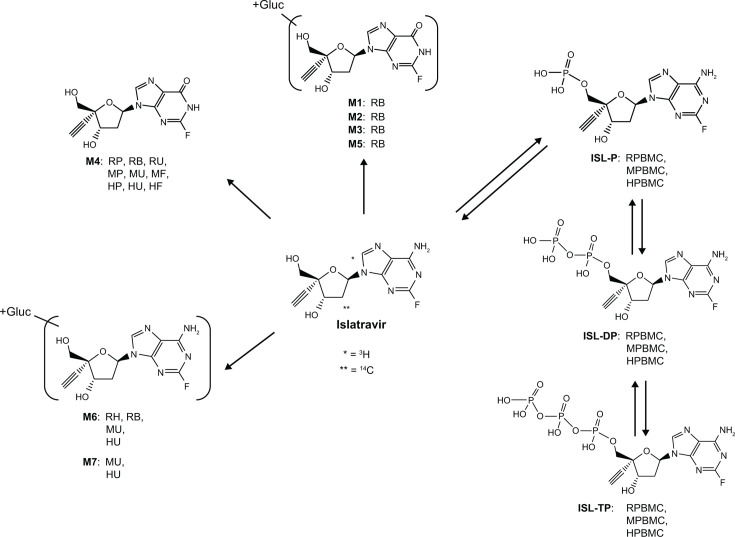 Fig 1
Fig 1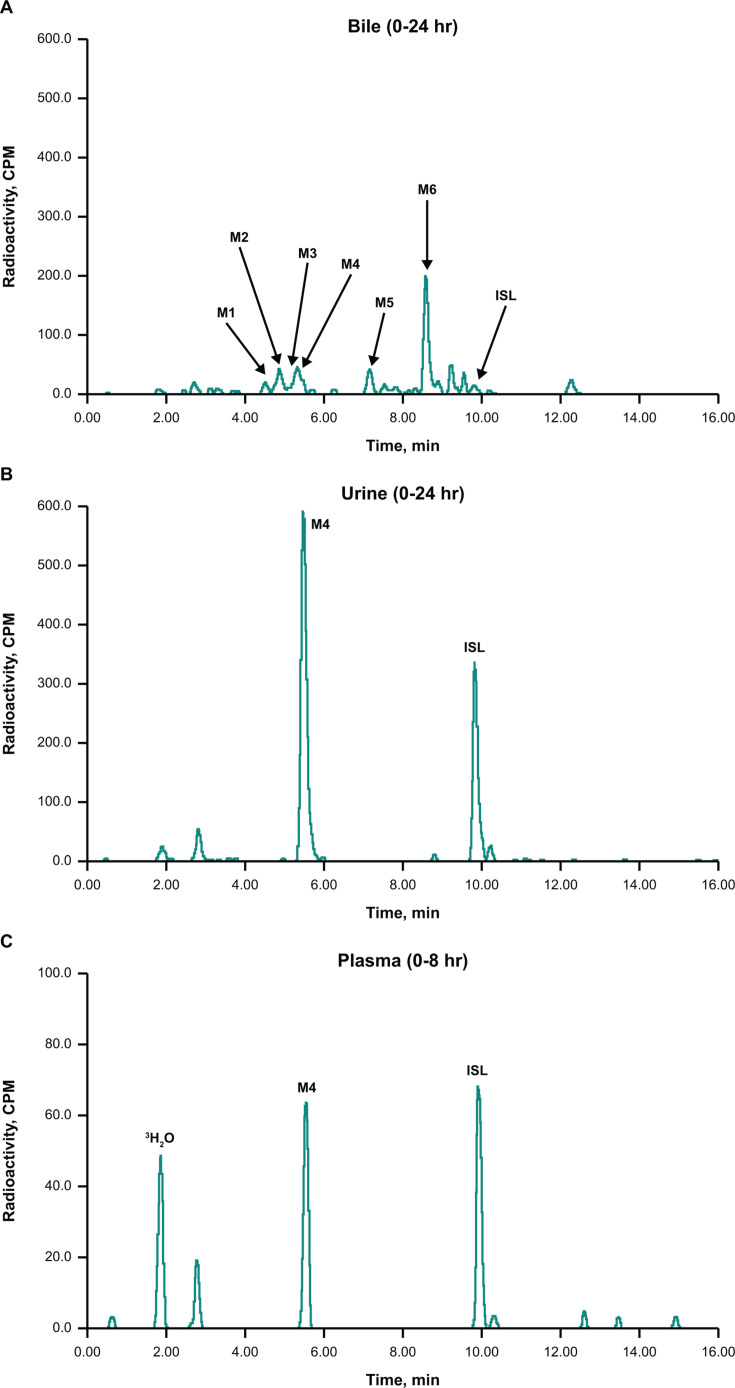 Fig 2
Fig 2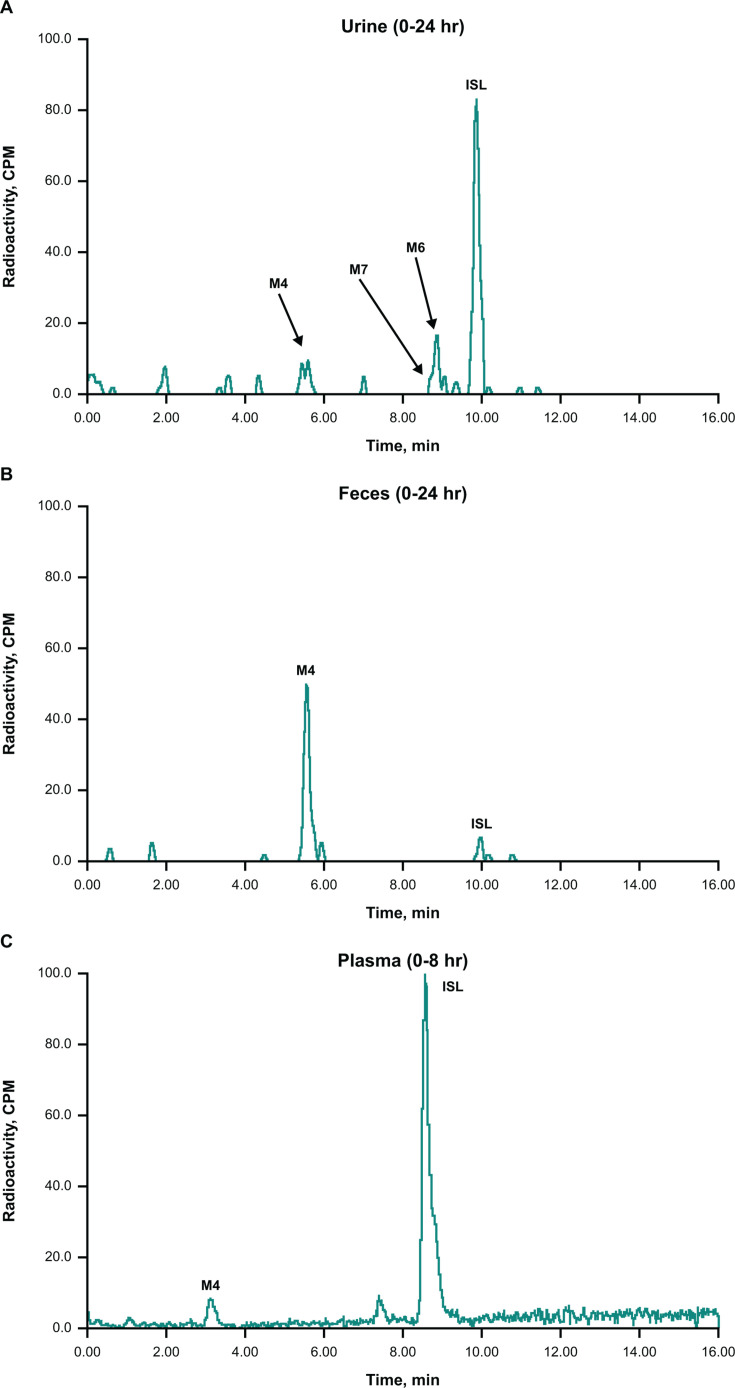 Fig 3
Fig 3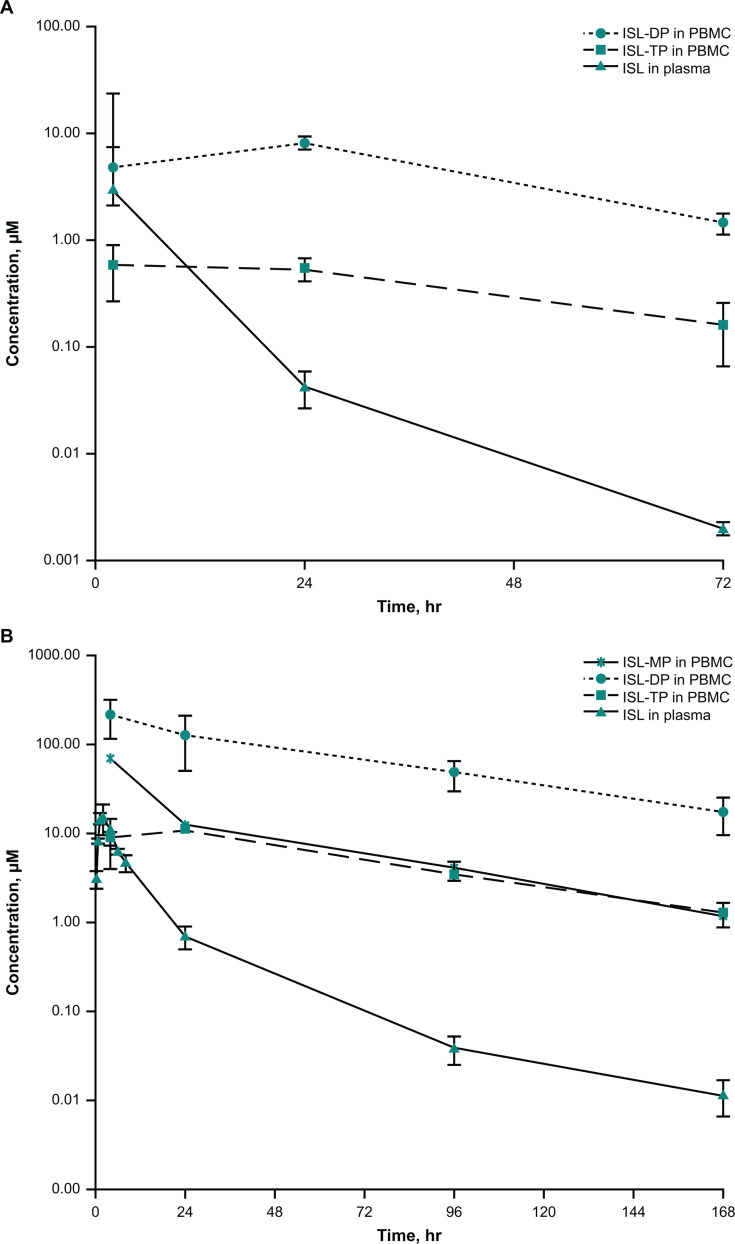 Fig 4
Fig 4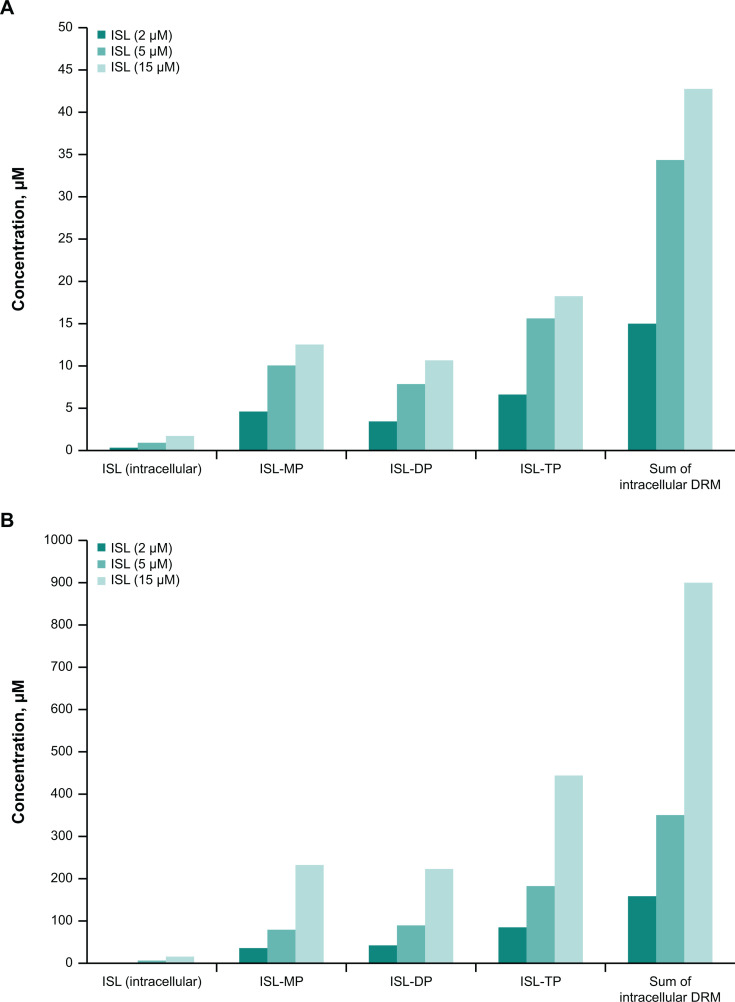 Fig 5
Fig 5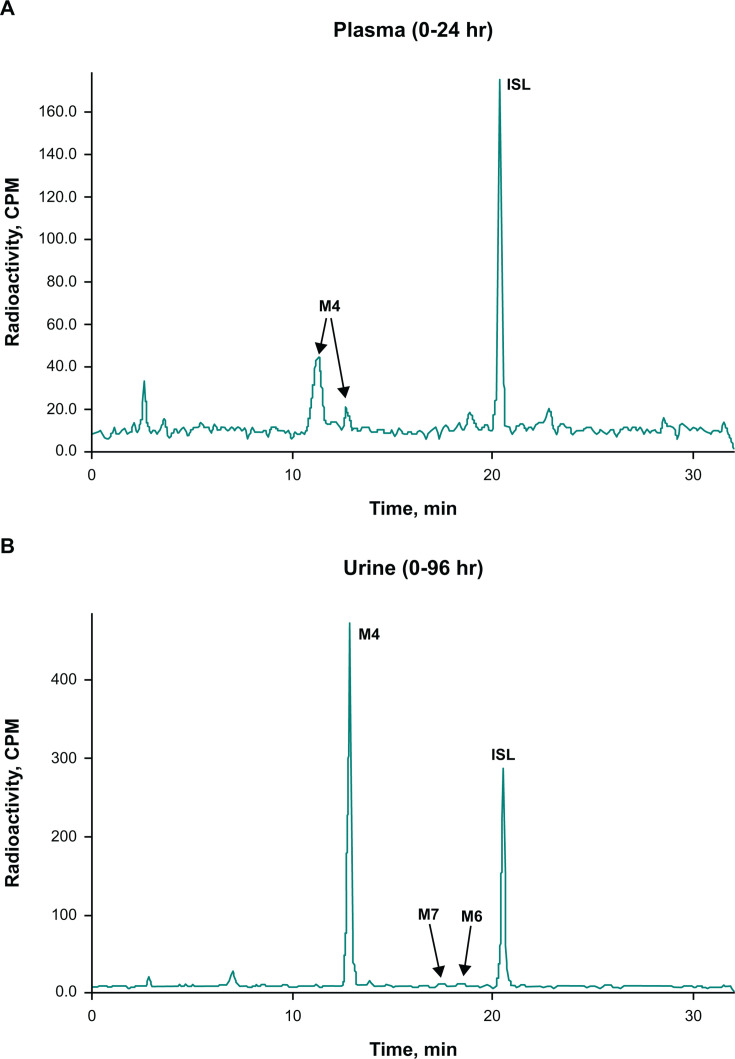 Fig 6
Fig 6| Animal model | Fraction of radioactivity recovery, mean % (SD) | ||||
|---|---|---|---|---|---|
| Urine | Feces | Bile | Cage wash/wipes | Total | |
| Rats ( | 73.7 (15.9) | 4.7 (1.0) | 8.3 (3.3) | 4.4 (4.4) | 91.1 (10.0) |
| Rhesus monkeys ( | 46.6 (8.7) | 31.0 (8.3) | 3.6 (1.7) | 1.2 (0.3) | 82.4 (2.4) |
| Component | Percentage of total radioactivity (dose percentage) | |||
|---|---|---|---|---|
| Plasma (0–8 hours) | Urine (0–24 hours) | Feces (0–24 hours) | Bile (0–24 hours) | |
| Wistar Hannover rats ( | ||||
| ISL | NQ | 22.3 | NA | 0.2 |
| M1 | NQ | ND | NA | 0.3 |
| M2 | ND | ND | NA | 0.7 |
| M3 | ND | ND | NA | 0.9 |
| M4 | NQ | 43.1 | NA | 0.1 |
| M5 | ND | ND | NA | 0.5 |
| M6 | ND | ND | NA | 2.6 |
| Rhesus monkeys ( | ||||
| ISL | NQ | 34.3 | 2.7 | NA |
| M4 | NQ | 5.2 | 28.3 | NA |
| M6 | ND | 4.8 | ND | NA |
| M7 | ND | 0.9 | ND | NA |
| Urine | Feces | Total | |
|---|---|---|---|
| Fraction of radioactivity recovery, mean % (95% CI) | 91.4 (87.9–95.0) | 6.3 (3.7–8.9) | 97.7 (96.4–99.0) |
| Component | Percentage of total radioactivity (dose percentage) | ||
|---|---|---|---|
| Plasma (0–24 hours) | Urine (0–96 hours) | Feces (0–144 hours) | |
| ISL | 58 | 35 (32.0) | ND |
| M4 | 31 | 58 (53.0) | NQ |
| M6 | ND | NQ | ND |
| M7 | ND | NQ | ND |
| Unidentified metabolites | <7 | 1 (0.9) | ND |
| Parameter | GM (95% CI) |
|---|---|
| ISL AUC0–24 | 0.675 (0.617–0.740) |
| Total radioactivity AUC0–24 | 2.29 (2.09–2.51) |
| ISL | 0.263 (0.217–0.319) |
| Total radioactivity | 0.739 (0.610–0.898) |
| ISL | 0.00368 (0.00331–0.00413) |
| Total radioactivity | 0.0339 (0.0305–0.0380) |
| ISL | 0.50 (0.50, 0.50) |
| Total radioactivity | 0.50 (0.50, 0.50) |
- —Merck (Merck & Co., Inc., Rahway, NJ, USA)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHIV/AIDS drug development and treatment · Drug Transport and Resistance Mechanisms · Hepatitis C virus research
INTRODUCTION
Antiretroviral therapy plays an important role in reducing the mortality and morbidity associated with HIV-1 infection, as well as reducing transmission rates (1). Several agents and fixed-dose combination regimens are available for the treatment of HIV infection; however, limitations to available therapies include safety and tolerability issues and the emergence of drug-resistant variants (2). Development of new and improved antiretroviral agents for HIV-1 infection, including those that allow for alternative dosing schedules, is therefore warranted.
Islatravir (ISL, MK-8591) is a deoxyadenosine analog under investigation for the treatment of HIV-1 in daily and weekly dosing regimens (3–6). The structure of ISL (Fig. 1) leads to inhibition of HIV-1 replication by multiple mechanisms of action, including reverse transcriptase translocation inhibition and delayed chain termination, which contribute to the high potency of ISL against HIV-1 (and drug-resistant variants) and its high barrier to resistance (5, 7–9).
Proposed metabolites of ISL in in vivo and in vitro systems. + Gluc, glucuronide conjunction; HF, human feces; HPBMC, human peripheral blood mononuclear cell; HU, human urine; ISL, islatravir; ISL-DP, ISL diphosphate; ISL-P, ISL monophosphate; ISL-TP, ISL triphosphate; MF, monkey feces; MP, monkey plasma; MPBMC, monkey peripheral blood mononuclear cell; MU, monkey urine; RB, rat bile; RP, rat plasma; RPBMC, rat peripheral blood mononuclear cell; RU, rat urine.
The efficacy of ISL has been assessed in treatment-naive adults with HIV as monotherapy (10) and in combination with doravirine, demonstrating effective viral suppression (6). Following oral administration in humans, ISL is rapidly absorbed; following cellular uptake, ISL is phosphorylated to its active form, islatravir-triphosphate (ISL-TP), which interferes with HIV reverse transcriptase activity (11).
ISL is eliminated as unchanged drug by renal excretion (12, 13), with nonclinical studies demonstrating that approximately 30%–60% of the ISL total plasma clearance is attributed to renal excretion (12). Results from in vitro studies suggest that ISL is not a substrate of renal transporters, and therefore, renal excretion of ISL is likely mediated primarily via glomerular filtration (12, 14). Nonclinical data demonstrate that 4′-ethynyl-2-fluoro-2′-deoxyinosine (M4) is the major metabolite of ISL (12), and M4 is also excreted in the urine in humans (12, 15).
Understanding the metabolism and disposition of new therapeutic agents is an integral part of the pharmacokinetic (PK) and safety evaluation of a drug. A comprehensive evaluation of ISL was performed to provide data on the absorption, metabolism, and excretion of ISL through mass balance and metabolite profiling studies in nonclinical species and adults without HIV-1 infection, using radiolabeled ISL.
RESULTS
Excretion of radioactivity in nonclinical species
Following an oral dose of [^3^H]ISL in rats, the average total radioactivity recovered in the excreta, within 72 hours, represented 91.1% of the administered dose (Table 1). Most of the radioactivity was recovered in urine (73.7%), with smaller amounts being recovered in bile and feces (8.3% and 4.7%, respectively). In rhesus monkeys, the average total radioactivity recovered in the excreta within 120 hours represented 82.4% of the administered dose. Much of the radioactivity was recovered in urine (46.6%), followed by feces (31.0%), with small amounts recovered in bile (3.6%; Table 1).
Metabolite profiles in nonclinical species
Metabolism via oxidative deamination was the primary mechanism of elimination in rats, in which M4 was the major metabolite in excreta and accounted for 43.1% of the administered dose (Table 2). The radioactivity recovered in bile and urine was mostly in the form of metabolites (Fig. 2A and B); however, in urine, a substantial portion of radioactivity was recovered as unchanged parent compound, accounting for 22.3% of the administered dose (Table 2). Glucuronide conjugates of M4 (M1, M2, M3, and M5) were minor metabolites in bile, each accounting for <1% of the administered dose (Table 2). A minor glucuronide of ISL (M6) was also detected in bile (Fig. 2A). In rat plasma, ISL, M1, and M4 were the only drug-related species identified by radiochemical and/or high-resolution mass spectrometry (HRMS) analysis in a pooled 0–8-hour plasma sample (Fig. 2C; Table 2).
Representative radiochromatograms of pooled (A) bile, (B) urine, and (C) plasma from Wistar Hannover rats dosed orally with 5 mg/kg [3H]ISL. CPM, counts per minute; LC-HRMS, liquid chromatography HRMS. All metabolites, except those that could not be detected by LC-HRMS due to their small quantities or low ionization potential, were identified based on radiochromatographic profile, exact mass, and fragmentation pattern.
In rhesus monkeys, ISL was the major species in urine and represented 34.3% of the administered dose. M4 and glucuronide conjugates of ISL, M6 and M7, were minor metabolites in urine (Fig. 3A; Table 2). In feces, M4 was the major metabolite (Fig. 3B), accounting for 28.3% of the administered radioactivity, and ISL was a minor component (Table 2). Bile was not profiled due to the relatively low recovery (3.6% of the administered radioactivity; Table 1) in this matrix. In monkey plasma, no discernable peaks were identified in the radiochemical analysis of a pooled 0–8-hour plasma sample; however, both ISL and M4 were identified by HRMS analysis in the same sample (Fig. 3C).
Representative radiochromatograms of pooled (A) urine and (B) feces and LC-HRMS extracted ion chromatogram of (C) pooled plasma from rhesus monkeys dosed orally with 5 mg/kg [3H]ISL. LC-HRMS, liquid chromatography HRMS. All metabolites, except those that could not be detected by LC-HRMS due to their small quantities or low ionization potential, were identified based on radiochromatographic profile, exact mass, and fragmentation pattern.
Plasma and intracellular PK in nonclinical species
The concentration-time profiles and mean PK parameter values for ISL in plasma and for ISL-mono-, di-, and triphosphates (ISL-MP, -DP, and -TP) in peripheral blood mononuclear cells (PBMCs) following oral administration to rats are shown in Fig. 4A and summarized in Table 3, respectively. Levels of ISL in plasma were measurable for ≤72 hours but were below the lower limit of quantification (LLOQ) by the 168-hour time point. ISL plasma concentrations were maximal at 2 hours postdose and declined in a biphasic manner until the 72-hour time point, with an approximate terminal half-life (t½) of 10 hours (Table 3). The intracellular concentration of ISL in rat PBMCs was measurable only at 2 hours postdose. Levels of ISL-MP in PBMCs were below the LLOQ for all time points. ISL-DP and ISL-TP in PBMCs were measurable for ≤72 hours but below the LLOQ at the 168-hour time point (Fig. 4A). The apparent intracellular levels of ISL-DP increased from 2 to 24 hours but declined through 72 hours, with a t½ of 20 hours. The apparent intracellular levels of ISL-TP were constant between 2 and 24 hours postdose but decreased over the 24–72-hour time frame, with a t½ of 25 hours (Fig. 4A; Table 3).
Concentration-time profile of ISL in plasma and ISL-MP, -DP, and -TP in PBMCs following oral administration of ISL in (A) rats and (B) monkeys. (Mean ± SD; n = 3). ISL-MP levels were below the lower limit of quantitation at all time points.
TABLE 3: Mean PK parameter values for ISL and ISL-MP, -DP, and -TP in plasma and PBMCs following oral administration of ISL in rats and monkeysa,f,g
The concentration-time profiles and mean PK parameter values for ISL in plasma and ISL-MP, -DP, and -TP in PBMCs following oral administration to monkeys are shown in Fig. 4B and summarized in Table 3. ISL exhibited rapid absorption: time to maximum concentration (Tmax) was 1.3 hours postdose; concentrations declined in a multiphasic manner, with a t½ of 41 hours, approaching the intracellular half-lives of the phosphorylated anabolites (Fig. 4B; Table 3). The intracellular concentration of ISL in PBMCs was only measurable at 4 hours postdose. The apparent intracellular levels of ISL-DP were significantly higher than those of ISL-MP and -TP (Table 3).
In vitro metabolism
In ex vivo blood incubation assays, the incubation time of 2 hours was deemed sufficient to establish equilibrium based on equivalent concentrations of intracellular ISL concentrations in PBMCs and extracellular ISL. In both monkey and human PBMCs, intracellular levels of total ISL drug-related material increased linearly with increasing extracellular ISL concentrations (Fig. 5A and B). Phosphorylation of ISL was efficient, and intracellular levels were in the order of ISL-TP > ISL-MP ~ ISL-DP >> ISL. The ratio of ISL-TP to ISL-DP was approximately 2:1. The uptake of ISL into human PBMCs was more efficient than in monkeys: levels of total ISL drug-related material after a 2-hour incubation were consistently 10-fold higher in human PBMCs than in monkey PBMCs, irrespective of the initial ISL concentrations (Fig. 5A and B).
Intracellular concentrations of ISL, ISL-MP, -DP, and -TP anabolites in PBMCs following incubation of ISL (2, 5, and 15 µM) in fresh (A) monkey and (B) human blood for 2 hours. DRM, drug-related material.
Clinical study demographics and safety
A total of six male participants in general good health without HIV infection were enrolled, and all six completed the study. The mean age was 38 years (range, 29–54 years) with a mean body mass index of 26.7 kg/m^2^ (range, 20.5–30.9 kg/m^2^). Four participants identified as Black, one as Asian, and one as White; none identified as Hispanic or Latino. No adverse events or discontinuations were reported. No findings of significance with respect to vital signs, electrocardiograms, or laboratory safety results were observed.
Excretion of radioactivity and metabolite profile in humans
Following oral administration of [^14^C]ISL in humans, 97.7% of the radioactive dose was recovered within 336 hours, with 91.4% recovered in urine and 6.3% recovered in feces (Table 4). In human plasma, ISL was the major circulating species (58% of the total radioactivity in plasma), with M4 as the major metabolite (31%; Table 5; Fig. 6A). In urine, most of the radioactivity was assigned to M4 (Fig. 6B), accounting for 53% of the administered dose (Table 5), while unchanged ISL accounted for 32% of the dose (Fig. 6B; Table 5). Glucuronide conjugates of ISL, M6 and M7, were observed in trace quantities in urine.
Representative radiochromatograms of pooled (A) plasma and (B) urine following single-dose administration of 10 mg [14C]ISL in healthy male participants. AUC0–24, area under the concentration-time curve from 0 to 24 hours; CPM, counts per minute. Pooled plasma (0–24 hours in proportion to AUC0–24). In plasma, the main ISL metabolite, M4, was observed as two peaks; the splitting was attributed to a matrix effect. In urine, ISL metabolites M6 and M7 were detected by HRMS analysis only.
Plasma and intracellular PK in humans
In humans, following a single oral dose of ISL, median plasma Tmax was 0.50 hours for both ISL and total radioactivity (Table 6). Due to limitations of the bioanalytical assay, the ISL and radioactivity terminal elimination phases could not be characterized; therefore, area under the plasma concentration-time curve from 0 to infinity (AUC_0–∞_) and t½ are not reported.
DISCUSSION
ISL is an investigational deoxyadenosine analog with promising clinical efficacy data that shows potential in the treatment of HIV-1 (3, 4, 6, 10). Due to the importance of fully understanding the metabolic profile and disposition of new agents, here, we have characterized the absorption, metabolism, and excretion of ISL in nonclinical studies and a clinical study.
ISL was readily absorbed in all species following oral administration, as demonstrated by the radioactivity recovered in urine in humans and in urine and bile in rats and monkeys. Consistent with the exclusive recovery of ISL-related radioactivity in the urine in humans, the highest recovery of radioactivity in urine was seen in humans. The urine recovery was lower in rats, followed by monkeys. Parent ISL and metabolite M4 were the primary circulating ISL drug-related components and were prominently seen in urine and feces in all species.
In human plasma, ISL accounted for 58% of the circulating radioactivity, with M4 accounting for 31% of the radioactivity in plasma. In human urine, M4 accounted for the majority of the administered dose (53%), followed by ISL (32%). With respect to the administered dose, the relative amounts of ISL and M4 recovered in excreta varied across species. Fecal elimination of drug components was more prominent in monkeys vs rats or humans, although urinary elimination of the radioactive dose over fecal elimination was still dominant in monkeys. The proportion of M4 in monkey urine was also less than that in humans and rats, with M4 being the primary drug-related component in monkey feces. The presence of M4 in monkey feces indicated good absorption and suggested significant gut metabolism in this species, in which M4 is formed by oxidative deamination by adenosine deaminase (12), an enzyme with high expression in the gut (16, 17). The presence of ISL and M4 in excreta from all evaluated species indicated that ISL was eliminated primarily by a combination of renal excretion of unchanged drug and oxidative deamination to metabolite M4. In addition to M4, other ISL metabolites were observed, which include glucuronide conjugates of M4, as well as minor ISL glucuronide conjugates (M6 and M7; Fig. 1). However, most minor metabolites were present only in trace amounts (Fig. 1).
Antiviral activity of ISL is achieved through uptake into infected cells with subsequent phosphorylation to ISL-TP. Cellular uptake is assessed through measurement of the phosphorylated species in PBMCs. The PK of the ISL phosphorylated forms (ISL-MP, -DP, and -TP) was directly assessed in rats and monkeys; assessment of PBMCs in the clinical study was not feasible due to the challenges of isolating radiolabeled PBMCs. Efficient intracellular conversion of the parent drug was readily observed; however, quantitative assessment is limited owing to insufficient stabilization of the PBMC samples for both the rat and monkey studies. It was observed during method development (data not shown) that, without sample stabilization, ISL-TP hydrolyzes to the more stable ISL-DP form during sample storage. Therefore, it is likely that for these initial rat and monkey studies, ISL-TP hydrolyzed during sample storage resulting in an underestimation of ISL-TP concentrations and an overestimation of ISL-DP levels. Uptake and conversion were additionally assessed in vitro via direct incubation of ISL in PBMCs (monkey and human). ISL cellular uptake was seen, with efficient phosphorylation to the mono-, di-, and triphosphate forms. The active ISL-TP form was most prominent. The extent of ISL uptake into human PBMCs was more efficient than in monkey PBMCs; levels of total ISL drug-related material were consistently 10-fold higher in human PBMCs than in monkey PBMCs, irrespective of the initial ISL concentration. Comparison of the monkey and human results suggests a potential species difference in intracellular ISL uptake and is consistent with previous reports (18). This species difference may pose challenges when using monkeys as nonclinical species to predict ISL and ISL-TP concentrations in humans.
These studies characterized the absorption, metabolism, and elimination of ISL; however, limitations are acknowledged. Sample stability issues limited the profiling of the ISL-MP, -DP, and -TP intracellular PK in rats and monkeys. Assay sensitivity yielded issues with monkey plasma radioactivity assessments and with the human ISL PK and radioactivity assessments. It is also acknowledged that the nonclinical and clinical studies had small sample sizes with only single-dose administration of ISL. Based on multiple-dose ISL PK data, single-dose administration should be predictive of multiple-dose behavior (3). In the human study, the geometric mean ratio of ISL to total radioactivity could not be effectively evaluated because data older than 24 hours were not evaluable due to the limitation of the bioanalytical assay. These data would have provided further evidence regarding the degree of circulating parent ISL.
Across nonclinical species and humans, ISL is well absorbed following oral administration. ISL and the metabolite M4 are the major circulating drug components across species, and ISL and M4 are the primary drug-related components in excreta. ISL is readily taken up cellularly, with efficient phosphorylation to the mono-, di-, and triphosphate forms. The pharmacologically active ISL-TP is the primary intracellular phosphorylated species. Clinical development of oral once-monthly ISL for HIV pre-exposure prophylaxis has been discontinued due to dose-dependent decreases in total lymphocyte and CD4+ T-cell counts seen across the ISL development program (19, 20). However, ISL continues to be investigated as part of a two-drug HIV-1 treatment regimen with doravirine (at a dose of 0.25 mg once daily; NCT05631093) and with lenacapavir (at a dose of 2 mg once weekly; NCT05052996) (20, 21). These lower doses of ISL have not been associated with decreases in total lymphocyte or CD4+ T-cell counts. This characterization of the absorption, metabolism, and elimination of ISL supports the further development of ISL for the treatment of HIV-1.
MATERIALS AND METHODS
Chemicals
For the nonclinical rat and monkey excretion and mass balance study, [^3^H]ISL was synthesized by Process Chemistry, Merck & Co., Inc., Rahway, NJ, USA, with a specific activity of 2.464 µCi^−1^ (0.7228 Ci^−1^) and radiochemical purity of 97%. For the rat excretion and mass balance study, unlabeled ISL was combined with [^3^H]ISL in a solution of [^3^H]ISL with a nominal concentration of 1.0 mg/mL and a measured specific activity of approximately 72.5 µCi/g. For monkeys, the oral dosing solution (1.0 mg/mL) had a nominal specific activity of approximately 1.4 µCi/g. For the human excretion and mass balance study, [^14^C]ISL was synthesized by Analytical Research and Development, Merck & Co., Inc., Rahway, NJ, USA and was supplied as an oral suspension (10 mg/mL) with a specific activity of approximately 6.2 µCi/mg (0.002 µCi^−1^) and radiochemical purity of 100%.
In vivo animal studies
All animal studies were conducted using protocols in accordance with the Institutional Animal Care and Use Committee at Merck & Co., Inc., Rahway, NJ, USA, which adhere to the regulations outlined in the US Department of Agriculture Animal Welfare Act.
Nonclinical assessment of ISL radioactivity mass balance, metabolic profiling, and PK assessments was performed on rats and monkeys. Bile duct-cannulated male Wistar Hannover rats (n = 3) weighing 300–360 g and intact male Wistar Hannover rats (n = 3) weighing 280–300 g with cannulated jugular veins were fasted overnight then dosed with 5 mL/kg [^3^H]ISL (5 mg/kg; approximately 100 µCi per animal). Bile, urine, and fecal samples were collected from the bile duct-cannulated animals, and blood was collected from the intact animals through 72 hours postdose; cages were rinsed and wiped for radioactivity analysis at the end of the study. Bile duct-cannulated male rhesus monkeys (n = 3) weighing 6.9–9.2 kg were fasted overnight then dosed via oral gavage with 5 mL/kg [^3^H]ISL (5 mg/kg; approximately 60 µCi per animal). Bile was collected through 72 hours postdose; blood samples were collected through 96 hours postdose; and urine and fecal samples were collected through 120 hours postdose. Cages were rinsed at intervals up to 120 hours postdose.
In vivo metabolism studies
For PBMC distribution and anabolism assessment in rats and monkeys, male Wistar Hannover rats (n = 12) weighing approximately 300 g and male rhesus monkeys (n = 3) weighing 4.5–5.0 kg were given an oral suspension of 5 mL/kg ISL at final doses of 30 mg/kg and 50 mg/kg, respectively. Blood samples were collected for plasma and PBMC analysis through 168 hours postdose.
In vitro metabolism studies
Fresh heparinized human and monkey blood samples were collected from at least three donors (each species). Blood was incubated with ISL (2, 5, and 15 µM) at 37°C for 2 hours. After incubation, PBMCs were isolated and analyzed for intracellular levels of ISL, ISL-MP, -DP, and -TP.
Clinical study
Study participants (n = 6) were healthy adult males aged 18–55 years with a body mass index ≥18 and ≤32 kg/m^2^. All participants signed written informed consent before study entry. Participants were enrolled between 26 February 2020 and 26 May 2021. Participants received a single oral dose of 10 mg (approximately 62 µCi) [^14^C]ISL in the fasted state. Whole blood, plasma, urine, fecal samples, and toilet tissue were collected throughout the study for radioactivity mass balance, metabolic profiling, and PK profiling. Blood was collected through 120 hours postdose; urine and fecal samples were collected through 336 hours postdose. Clinical safety was monitored.
Measurement of radioactivity
All samples collected in the animal and human metabolic profiling studies were analyzed for total radioactivity content by liquid scintillation counting. Actual doses administered in disintegrations per minute were used to calculate the percentage of dose recovered.
LC-HRMS and radiochromatographic analysis
Aliquots of urine were pooled proportionally relative to the total volume collected over 24 hours for rats and monkeys and pooled over 96 hours for humans. After centrifugation, the resulting supernatant was analyzed by liquid chromatography (LC)-HRMS with radiometric detection.
For rats and monkeys, bile samples from the 0- to 24-hour collection interval were pooled, mixed with acetonitrile, vortexed, and sonicated. After centrifugation, the resulting supernatants were analyzed by radiometric LC-HRMS. Fecal homogenate samples from 0 to 24 hours were pooled. Aliquots of fecal homogenate were mixed with methanol, vortexed, and centrifuged, and the resulting supernatant was analyzed by radiometric LC-HRMS. Recovery of radioactivity in the reconstituted samples was measured using liquid scintillation counting.
For humans, aliquots of fecal samples were pooled proportionally relative to weight across individuals to generate a single fecal homogenate. The sample was mixed with acetonitrile, vortexed, and centrifuged, and the resulting supernatant was analyzed by LC-HRMS with offline radiometric profiling.
Plasma samples from 0 to 8 hours in rats and monkeys and 0 to 24 hours for humans were pooled for each species according to the Hamilton time proportional pooling algorithm (22) to yield one sample that had a concentration proportional to the PK AUC. An aliquot of the pooled sample was treated by protein precipitation with the addition of acetonitrile or acetonitrile plus methanol [90/10 (vol/vol)] followed by vortex mixing and centrifugation. The resulting supernatants were analyzed by radiometric LC-HRMS. The LLOQ for radiometric detection was approximately 10–20 counts per minute.
Analysis of islatravir in plasma
Following extraction with acetonitrile, the concentrations of ISL in rat and monkey plasma were determined by liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis with a dynamic range of 5–5,000 nM. Human plasma was extracted with methyl-tert-butyl ether, and ISL levels were determined using a validated LC-MS/MS method. The analytical range of this method was 0.0682–68.2 nM (20.0–20000.0 pg/mL).
Analysis of the intracellular levels of ISL and ISL-MP, -DP, and -TP in PBMCs
PBMCs were isolated from heparinized rat, monkey, and human blood samples by density gradient centrifugation. PBMCs collected from in vitro studies were stabilized with 70% methanol before freezing, whereas PBMCs collected from monkeys and rats in in vivo studies were not. The intracellular concentrations of ISL, ISL-MP, -DP, and -TP were determined by an LC-MS/MS assay, for which separation of the ISL phosphate anabolites was achieved by ion exchange chromatography. The LLOQ was between 2 and 13 nM depending on the volumes of cell suspension used in the assay. Analyte concentrations in cell samples across species were determined by weighted linear regression of the standard curve and normalized to the total viable PBMC cell counts. Concentrations in units of picomole per million PBMCs were converted to micromolar using a cell volume of 0.200 pL per PBMC (23, 24).
PK analysis
PK parameter values were calculated using noncompartmental methods in Watson LIMS (Thermo Fisher Scientific; Waltham, MA, USA) for the nonclinical studies and Phoenix WinNonlin version 8.1 (Certara; Princeton, NJ, USA) for the clinical study. The AUCs from 0 to a specific time, such as 24 hours, 72 hours, and so on (AUC_0–24_ and AUC_0–72_), were calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. The maximum plasma concentration, concentration at 24 hours, and Tmax were obtained by graphical inspection of the plasma concentration-time data. The t½ in nonclinical species was determined by selected time points based on visual inspection of the data.
Statistical analysis
For the clinical study, prior to analysis, individual values of each PK parameter and analyte were natural log-transformed and evaluated by use of a linear mixed-effects model with analyte (ISL and total radioactivity) as a fixed effect and participant as a random effect. Model-based summary statistics, geometric means, and 95% confidence intervals were calculated. Arithmetic means (on the raw scale) and corresponding 95% confidence intervals for recovery of total radioactivity (as a percentage of dose administered) were provided for urine and feces, as well as in total.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1WHO. 2023. Global HIV programme. Available from: https://www.who.int/teams/global-hiv-hepatitis-and-stis-programmes/hiv/treatment#:~:text=The%20main%20objective%20of%20treatment,life%20and%20preventing%20HIV%20transmission. Retrieved 2 Jan 2025.
- 2DHHS. 2023. Department of health and human services. Panel on antiretroviral guidelines for adults and adolescents guidelines for the use of antiretroviral agents in adults and adolescents with HIV. Available from: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/adult-adolescent-arv/guidelines-adult-adolescent-arv.pdf. Retrieved 2 Jan 2025.
- 3Matthews RP, Ankrom W, Friedman E, Jackson Rudd D, Liu Y, Mogg R, Panebianco D, De Lepeleire I, Petkova M, Grobler JA, Stoch SA, Iwamoto M. 2021. Safety, tolerability, and pharmacokinetics of single- and multiple-dose administration of islatravir (MK-8591) in adults without HIV. Clin Transl Sci 14:1935–1944. doi:10.1111/cts.1304834463432 PMC 8504818 · doi ↗ · pubmed ↗
- 4Molina JM, Yazdanpanah Y, Afani Saud A, Bettacchi C, Chahin Anania C, De Jesus E, Olsen Klopfer S, Grandhi A, Eves K, Robertson MN, Correll T, Hwang C, Hanna GJ, Sklar P. 2021. Islatravir in combination with doravirine for treatment-naive adults with HIV-1 infection receiving initial treatment with islatravir, doravirine, and lamivudine: a phase 2b, randomised, double-blind, dose-ranging trial. Lancet HIV 8:e 324–e 333. doi:10.1016/S 2352-3018(21)00021-734000227 · doi ↗ · pubmed ↗
- 5Diamond TL, Ngo W, Xu M, Goh SL, Rodriguez S, Lai MT, Asante-Appiah E, Grobler JA. 2022. Islatravir has a high barrier to resistance and exhibits a differentiated resistance profile from approved nucleoside reverse transcriptase inhibitors (NRT Is). Antimicrob Agents Chemother 66:e 0013322. doi:10.1128/aac.00133-2235546110 PMC 9211433 · doi ↗ · pubmed ↗
- 6Molina JM, Yazdanpanah Y, Afani Saud A, Bettacchi C, Chahin Anania C, Klopfer SO, Grandhi A, Eves K, Hepler D, Robertson MN, Hwang C, Hanna GJ, Correll T. 2022. Brief report: efficacy and safety of oral islatravir once daily in combination with doravirine through 96 weeks for treatment-naive adults with HIV-1 infection receiving initial treatment with islatravir, doravirine, and lamivudine. J Acquir Immune Defic Syndr 91:68–72. doi:10.1097/QAI.000000000000287935972855 PMC 9377497 · doi ↗ · pubmed ↗
- 7Kawamoto A, Kodama E, Sarafianos SG, Sakagami Y, Kohgo S, Kitano K, Ashida N, Iwai Y, Hayakawa H, Nakata H, Mitsuya H, Arnold E, Matsuoka M. 2008. 2’-deoxy-4’-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int J Biochem Cell Biol 40:2410–2420. doi:10.1016/j.biocel.2008.04.00718487070 · doi ↗ · pubmed ↗
- 8Salie ZL, Kirby KA, Michailidis E, Marchand B, Singh K, Rohan LC, Kodama EN, Mitsuya H, Parniak MA, Sarafianos SG. 2016. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4’-ethynyl-2-fluoro-2’-deoxyadenosine (E Fd A). Proc Natl Acad Sci U S A 113:9274–9279. doi:10.1073/pnas.160522311327489345 PMC 4995989 · doi ↗ · pubmed ↗