Exploration of the Fusidic Acid Structure Activity Space for Antibiotic Activity
Yoon-Suk Kang, Simone C. Silva, Kenneth Smith, Krissty Sumida, Yuhan Wang, Lucius Chiaraviglio, Ramachandra Reddy Donthiri, Alhanouf Z. Aljahdali, James E. Kirby, George A. O’Doherty

TL;DR
This study explores how modifying the structure of fusidic acid can improve its antibiotic activity, particularly against Gram-positive bacteria.
Contribution
The study identifies specific structural modifications to fusidic acid that retain or enhance its antibacterial activity.
Findings
Modifications to the A-ring alcohol of fusidic acid retained modest activity against Gram-positive bacteria.
Side-chain carboxylic acid esters and C-ring oxidations were ineffective or poorly tolerated.
The fusidic acid pyrazine-2-carboxylate may act as a pro-drug based on activity differences in assays.
Abstract
Fusidic acid is a translation inhibitor with activity against major Gram-positive bacterial pathogens such as S. aureus. However, its activity against Gram-negatives is poor based on an inability to access its cytoplasmic target in these organisms. Opportunities for functionalization of the fusidic acid scaffold to enhance activity against Gram-negative pathogens have not been explored. Using an activity-guided synthetic strategy, the tolerance of the tetracyclic natural product to derivatization at the A- and C-rings and its carboxylic acid side chain was explored with the goal of enhancing its activity spectrum and pharmacological properties. All side-chain carboxylic acid esters were inactive. Oxidation of the C-ring alcohol and oxime were not tolerated either. A number of esters of the A-ring alcohol retained modest activity against Gram-positive bacteria and were informative for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
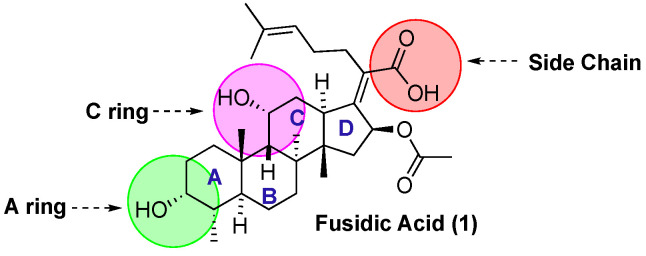 Figure 1
Figure 1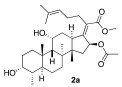 Figure 2
Figure 2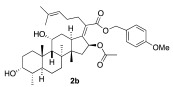 Figure 3
Figure 3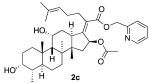 Figure 4
Figure 4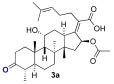 Figure 5
Figure 5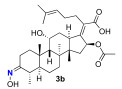 Figure 6
Figure 6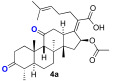 Figure 7
Figure 7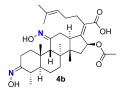 Figure 8
Figure 8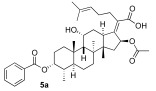 Figure 9
Figure 9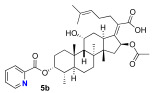 Figure 10
Figure 10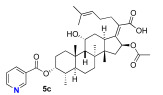 Figure 11
Figure 11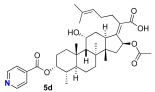 Figure 12
Figure 12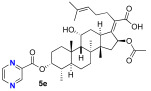 Figure 13
Figure 13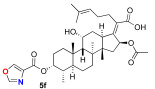 Figure 14
Figure 14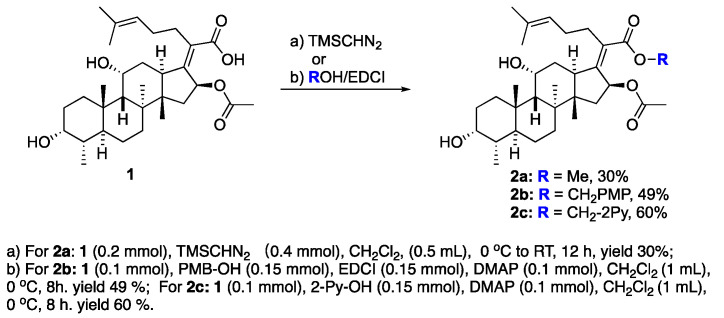 Figure 15
Figure 15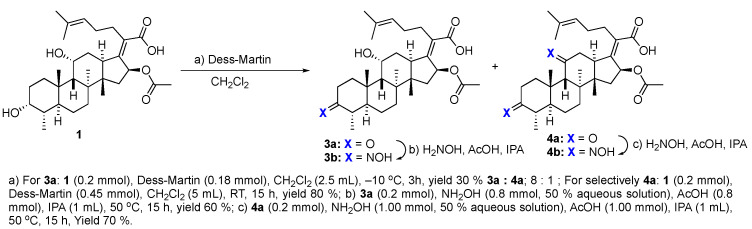 Figure 16
Figure 16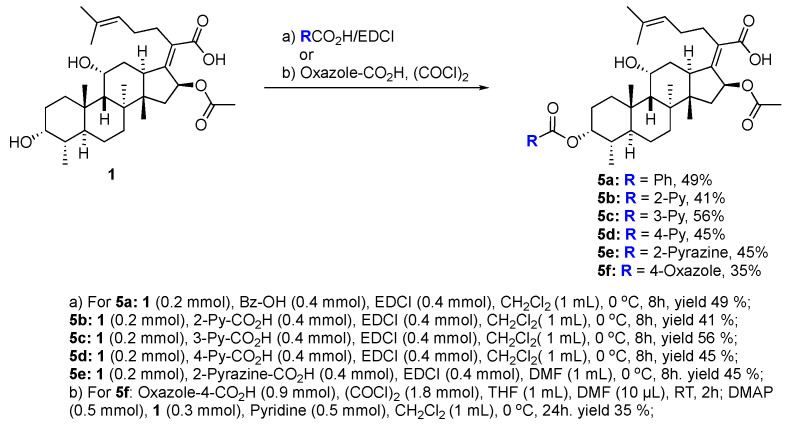 Figure 17
Figure 17- —NSF
- —NIH
- —Massachusetts Life Sciences Center
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhenothiazines and Benzothiazines Synthesis and Activities · Microbial Natural Products and Biosynthesis · Synthesis and biological activity
1. Introduction
Fusidic acid is a steroid antimicrobial natural product with potent activity against methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible Staphylococcus aureus (MSSA), β-hemolytic streptococci; potent in vitro activity against Neisseria gonorrhoeae; and in vitro and in vivo activity against extensively drug-resistant Mycobacterium tuberculosis [1,2,3]. There is clearly a great need for new Gram-negative antibiotics that can reduce selective resistance pressure through diversification of our antimicrobial therapeutic portfolio [4]. Our approach to the problem of antibiotic resistance centers on the derivatization of existing antibiotic natural products [5,6]. The drug is water-soluble, stable (shelf life 3 years), and highly orally available, and therefore can be given intravenously, orally, or used topically [7]. It has an excellent safety profile [8,9], a long half-life in humans, and is eliminated through biliary excretion. Fusidic acid concentrates well in all tissues; however, it does not cross the blood–brain barrier.
Fusidic acid targets ribosome translocation by inhibiting function of the ribosome-associated protein elongation factor G (EF-G), which assists in the translocation of transfer RNA in the ribosome. It achieved primary endpoints in a phase III US clinical trial (i.e., non-inferiority to linezolid in acute bacterial skin and skin structure infections) [1]. It is generally considered a static agent, but killing of Staphylococcus aureus and especially Streptococcus pyogenes is evident in hollow-fiber infection models [10]. Resistance to fusidic acid has been found in S. aureus [11] as a result of single-step mutations in fusidic acid’s ribosomal target, EF-G (encoded by the gene fusA), conferring high-level resistance, and in the ribosomal protein RplF (encoded by the gene fusE). Although multiple mutations can be isolated in the laboratory, only a minority (e.g., organisms encoded with fusA, L461K, and to a lesser extent H457Y and H457Q) are observed in clinical isolates and at low frequency, suggesting a significant fitness cost, supported by in vitro and in vivo data, including human colonization experiments [12,13,14,15]. Higher loading dose regimens have further addressed concern about selection of fusA resistance during monotherapy [1]. Plasmid-mediated, low-level resistance conferred by the FusB family proteins are the predominant resistance mechanism in Gram-positive clinical isolates. These proteins bind to EF-G and overcome the inhibitory effect of fusidic acid [16,17,18].
Brennan-Krohn et al. recently described fusidic acid’s potent and broad-spectrum activity against highly resistant, carbapenemase-producing Enterobacterales (CRE) when combined with colistin, which is known to permeabilize the outer Gram-negative bacterial membrane [19]. This co-dosing effect proved to be true even in colistin-resistant strains. We further note its potent activity against intracellular Legionella pneumophila [20] and facultative intracellular Brucella neotomae (unpublished). Others have also identified fusidic acid as having compelling axenic activity against L pneumophila when grown axenically [21,22]. These fastidious organisms have a lower permeation barrier to prototypical Gram-positive agents. Potent activity is also noted in Escherichia coli (E. coli) mutants with increased permeation (i.e., ΔtolC or lptD4213 genetic mutations) [23,24]. Notably, polymyxin B nonapeptide, an outer membrane permeabilization agent without direct antimicrobial activity, confers drastically reduced fusidic acid MICs for Klebsiella pneumonia, Acinetobacter baumannii, and Pseudomonas aeruginosa into the 1 μg/mL range [25]. Notably, FusB, and presumably related FusB family proteins, do not bind to E. coli EF-G and fail to overcome fusidic acid inhibition of E. coli in vitro translation systems [26,27]. Taken together, these results suggest that fusidic acid would have potent Gram-negative activity if it could penetrate the outer Gram-negative membrane, as there are no known pre-existing resistance mechanisms circulating in these pathogens [28].
As part of a long-standing interest in natural product medicinal chemistry, we became interested in the structural space around fusidic acid [29,30,31,32,33]. In particular, we wanted to explore whether the antibacterial activity of fusidic acid could be enhanced through derivatization. The fusidic acid scaffold has been previously investigated by others [34,35], most notably investigating the anti-malarial structure–activity relationship (SAR) [36,37]. A series of analogues modified at C-21 showed improved potency against P. falciparum [38]. Other have also explored modification of the A- and C-ring alcohols (Figure 1) [39,40,41].
2. Results and Discussion
Our own SAR efforts were aimed at identifying sites that showed promise of systematic variation compatible with generation and screening of large libraries of compounds. Specifically, we looked to identify which portions of the tetracyclic steroidal core of fusidic acid were amenable to functionalization without disrupting its potency as an antibacterial agent. The knowledge gained from this effort would in turn aid future studies aimed at expanding its antimicrobial activity spectrum. Initial efforts aimed at the de novo design using molecular modeling/docking studies of potential analogues was limited by the difficulty in finding a static structure that replicated the known X-ray and EPR structures of fusidic acid bound to EF-G–ribosome complexes [39,40,41,42,43,44]. As a result, we settled on the use of an activity-guided synthetic/screening strategy. For the screening, we chose to use S. aureus as the Gram-positive model organism and three types of E. coli: a wild type (E. coli wt), an E. coli with its TolC efflux transporter protein knocked out (E. coli ΔtolC) [45], and an E. coli mutant with a hyperpermeable outer membrane due to defective lipopolysaccharide transport (E. coli LptD4213) [46]. An E. coli-based in vitro transcription-translation assay was also used to validate that any of the newly discovered analogues with anti-S. aureus or anti-E. coli activity were targeting the ribosome [24]. At the outset, we identified three sites on the molecule that could be easily derivatized (Figure 1): the side-chain carboxylic acid (esters), the A ring C-3 alcohol (ketone, oxime and esters), and the C ring C-11 alcohol (ketone and oxime). Herein, we report the results of this effort.
The preparation of side-chain ester analogues began with the straightforward conversion of fusidic acid (1) into carboxylic acid esters. Specifically, we chose the smallest possible methyl ester, 2a, as well as the larger electronic rich para-methoxybenzyl ester 2b and electron-deficient 2-pyridylmethylester 2c with a basic nitrogen (Scheme 1). The known methyl ester 2a was easily prepared upon treatment with excess TMS-diazomethane in CH_2_Cl_2_, affording 2a with a 30% yield. In this case, the diol functionality in fusidic acid alleviated the need for alcohol solvents [47]. The other two esters, 2b and 2c, were prepared using carbodiimide coupling chemistry, specifically 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI). The selectivity issues associated with the fact that the fusidic acid structure contains both a carboxylic acid and two secondary alcohols were easily addressed by performing the coupling with a slight excess of alcohol (1.5 equiv), affording 2b and 2c with 49% and 60% yields, respectively. Because we prioritized purity over yield, the yields for these transformations were not optimized. Three purified esters were screened for antibiotic activity against S. aureus, and either a wild type E. coli K-12 strain or E. coli strains with increased permeability characteristics (ΔtolC and lptD4213 mutants) [48]. Unfortunately, no activity was found for the three different esters 2a–c (Table 1). Interestingly, this contrasted with prior reports of somewhat better activity for methyl ester 2a and a small subset of additional side-chain esters [41]. Therefore, we turned our efforts toward the generation of analogues that were functionalized on the A-ring and C-ring.
We next explored the effects on antibacterial activity of A- and C-ring substitution (Scheme 2). To accomplish this, we searched for reaction chemistry that selectively reacted at the A-ring alcohol over the alcohol in the C-ring and vice versa. We first explored oxidation chemistry to selectively access a mono-ketone in the A- or C-ring. For steric reasons, it is reasonable to assume that the A-ring alcohol would be more accessible to reagent over the C-ring. To some extent, this proved to be true for the oxidation of fusidic acid with Dess–Martin periodate. That is to say, the A-ring alcohol was the more reactive, although it was difficult to prevent overoxidation to the diketone. Thus, under our optimized conditions, a 30% yield of fusidic acid mono-ketone was prepared when fusidic acid 3a was reacted with 0.9 equivalent Dess–Martin reagent. In contrast, the further oxidized diketone 4a product was significantly easier to obtain when excess (2.5 equiv) Dess–Martin reagent was used (80% yield). To further diversify the fusidic acid structural motifs, ketones 3a and 4a were converted into oximes 3b and 4b. This was simply accomplished with mono-ketone 3a by exposing it to excess aqueous hydroxyamine (4 equiv). Under these conditions, the A-ring oxime 3b was able to be isolated with a 60% yield. Under similar conditions with a slightly higher amount of aqueous hydroxyamine (5 equiv), the bisketone 4a was able to be converted into bisoxime 4b with a 70% yield. The four analogues 3a/b and 4a/b were tested for their ability to inhibit growth of S. aureus and E. coli strains and for their ability to inhibit in vitro translation in an assay performed with E. coli S30 ribosome extracts (Table 2). In contrast to a complete block of antimicrobial activity observed with carboxylic acid esterification, the fusidic acid A-ring oxidation retained modest antimicrobial activity against S. aureus. Others have found similar activity for mono-ketone 3a and oxime 3b against S. aureus and mycobacteria [45,46]. As for fusidic acid, active analogues did not have observable activity in wild-type E. coli or its hyperpermeable lptD4213 mutant at the concentrations tested, but did have activity in the ΔtolC mutant, consistent with exclusion of fusidic acid and its analogues by efflux pump mechanisms. Activity in the E. coli in vitro translation assay (IC_50_) correlated with antimicrobial activity (MIC) against S. aureus, consistent with the ability of fusidic acid and active analogues to inhibit translation if they gain access to their ribosomal target. Unfortunately, further oxidation of the C-ring and/or oxime formation had a deleterious effect on activity.
We next decided to further explore substitution of the fusidic acid A-ring with ester formation with a series of aromatic carboxylic acids. The structure of the aromatic ring was varied by the inclusion of one to two basic nitrogen atoms in the ortho, meta, and para portion of the benzene ring. To accomplish this, we returned to the use of carbodiimide coupling chemistry. In contrast to the condition used for the selective coupling of the fusidic acid carboxylic acid with alcohols, we chose to first premix the EDCI and the desired carboxylic acid coupling partner before exposure to fusidic acid. Typically, the acylurea intermediate that is formed from the carboxylic acid–EDCI carboxylic acid/EDCI admixture was used in a two-fold excess to fusidic acid. Under these conditions, fusidic acid A-ring esters 5a–e were exclusively formed and able to be isolated, with yields ranging from 41% to 56% (Scheme 3). The six analogues 5a–f were screened for antibiotic activity (see Table 3).
While mostly inactive against E. coli, interesting activity was observed against S. aureus. This was especially true for ester 5e, the esterification product with pyrazine-2-carboxylic acid, which had an MIC of 0.5 μM. It appears both nitrogen atoms played a role in the activity of 5e, as when one or both nitrogen atoms were removed to form either 2-pyridyl 5b or 3-pyridyl 5c, activity was reduced (16 μM). In addition, when the nitrogen was moved to a 1,4-relationship to the ester carboxylate, as in 5d, the activity dropped significantly (64 μM). The oxazole analogue 5f, which also has an SP^2^ nitrogen atom in a 1,2-relationship to the carboxylation, had similar activity (16 μM). However, it should be noted that the second-most active compound against S. aureus was the benzoate 5a (2–4 μM) with both nitrogen atoms removed. Analogues 5a–f were also evaluated for inhibitor effects on in vitro translation. With the exception of the 4-pyridyl analogue 5d, all the compounds showed similar ribosome inhibitory activity. Interestingly, the analogue most active against S. aureus, 5e, did not have corresponding potency in in vitro translation assays. This suggested the possibility that the improved antibacterial activity of 5e might be due to the selective hydrolysis of the pyrazine-2-carboxylate to the more potent fusidic acid. The hydrolysis of the more reactive pyrazine-2-carboxylate ester in 5e could have been enzyme-promoted or occurred during the longer incubation time used during antibacterial testing. However, potential improvements in ability to access the cytoplasmic EF-G target relative to intrinsic potency cannot be ruled out.
3. Materials and Methods
3.1. General Methods
^1^H and ^13^C{1H} spectra were recorded on Bruker 500 and 126 MHz NMR spectrometers, see the Supplementary Materials Section. Chemical shifts were reported relative to internal tetramethylsilane (δ 0.00 ppm) or CDCl_3_ (δ 7.26 ppm) or CD_3_OD (δ 4.89 ppm) for ^1^H-NMR and CDCl_3_ (δ 77.1 ppm) or CD_3_OD (δ 49.15 ppm) for ^13^C{1H}-NMR. Optical rotations were measured with a digital polarimeter in the solvent specified. Infrared (IR) spectra were obtained on a Bruker FT-IR spectrometer. Flash column chromatography was performed on ICN reagent 60 (60–200 mesh) silica gel. Analytical thin-layer chromatography was performed with precoated glass-backed plates (K6F 60 Å, F254) and visualized by quenching of fluorescence and charring after treatment with cerium ammonium molybdate or potassium permanganate stain. R_f_ values were obtained by elution in the stated solvent ratios (v/v). Ether, tetrahydrofuran (THF), dimethyl formamide (DMF), methylene chloride (DCM), methanol (MeOH), ethyl acetate (EA), hexane, and triethylamine (TEA) were dried by passing through activated silica gel (8 × 14 mesh) column with nitrogen gas pressure. All commercial reagents were obtained from the Fisher Scientific (Waltham, MA, USA) used without purification unless otherwise noted. Air- and/or moisture-sensitive reactions were carried out under an atmosphere of argon/nitrogen using oven/flame-dried glassware and standard syringe/septa techniques.
3.2. Experimental Procedures
methyl(Z)-2-((3R,4S,5S,8S,9S,10S,11R,13R,14S,16S)-16-acetoxy-3,11-dihydroxy-4,8,10,14-tetramethylhexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoate (2a)
A solution was prepared by dissolving fusidic acid (100 mg, 0.194 mmol, 1 equiv) in DCM (0.5 mL) and was cooled to 0 °C. A (0.2 mL) solution of TMSCH_2_N_2_, made from a (2M solution in ether) was added slowly at 0 °C. The reaction was brought up to room temperature and stirred for 12 h, then directly loaded onto a silica gel column. Flash chromatography (30% ethyl acetate in hexanes) gave 2a (colorless solid 93 mg, 90%); R_f_ = 0.58 (60% ethyl acetate/hexanes):
^1^H NMR (500 MHz, CDCl_3_) δ 5.84 (d, J = 8.4 Hz, 1H), 5.08 (tt, J = 7.2, 1.6 Hz, 1H), 4.34 (q, J = 2.7 Hz, 1H), 3.75 (q, J = 2.8 Hz, 1H), 3.63 (s, 3H), 3.05–3.01 (m, 1H), 2.53–2.36 (m, 2H), 2.30 (dt, J = 13.2, 3.3 Hz, 1H), 2.18 (dd, J = 13.0, 4.2 Hz, 1H), 2.11 (ddd, J = 16.5, 11.6, 5.4 Hz, 2H), 2.06–1.98 (m, 1H), 1.97 (s, 3H), 1.91–1.70 (m, 4H), 1.66 (d, J = 1.5 Hz, 3H), 1.62–1.56 (m, 1H), 1.59 (s, 5H), 1.55 (d, J = 2.0 Hz, 1H), 1.51 (ddd, J = 12.6, 4.5, 2.7 Hz, 1H), 1.37 (s, 3H), 1.26 (d, J = 14.2 Hz, 1H), 1.10 (dddd, J = 26.5, 12.2, 8.5, 5.9 Hz, 2H), 0.97 (s, 3H), 0.91 (d, J = 6.8 Hz, 3H), 0.90 (s, 3H).
^13^C NMR (126 MHz, CDCl_3_) δ 170.9, 170.5, 148.3, 132.7, 130.5, 123.2, 77.4, 74.6, 71.5, 68.4, 51.5, 49.3, 48.8, 44.0, 39.6, 39.2, 37.2, 36.3, 35.7, 32.5, 30.4, 30.1, 29.1, 28.4, 25.9, 24.3, 22.9, 21.1, 20.9, 17.9, 17.9, 16.1.
Spectral data for 2a matched those reported in the literature [49].
4-methoxybenzyl(Z)-2-((3R,4S,5S,8S,9S,10S,11R,13R,14S,16S)-16-acetoxy-3,11-dihydroxy-4,8,10,14-tetramethylhexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoate (2b)
4-Methoxybenzyl alcohol (21 mg, 0.15 mmol, 1.5 equiv), EDCI (28 mg, 0.15 mmol, 1.5 equiv), and DMAP (12 mg, 0.1 mmol, 1 equiv) were added to a solution of fusidic acid (50 mg, 0.1 mmol, 1.0 equiv) in anhydrous DCM (1 mL). The reaction mixture was stirred at 25 °C under a N_2_ atmosphere. After the reaction was determined to be complete by TLC (24 h), the reaction mixture was diluted with ethyl acetate (10 mL), washed with H_2_O (5 mL), and saturated with aqueous brine (5 mL). The organic layer was then dried over anhydrous MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography (27.5% EA in hexane) on silica gel to afford the product 2b (white solid, 31 mg, 49%). R_f_ = 0.5 (50% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 7.29 (dd, J = 8.5, 1.9 Hz, 2H), 6.87 (dd, J = 8.6, 1.9 Hz, 2H), 5.86 (d, J = 8.3 Hz, 1H), 5.13 (dd, J = 11.8, 1.7 Hz, 1H), 5.06–5.03 (m, 1H), 4.85 (dd, J = 11.9, 1.7 Hz, 1H), 4.32 (d, J = 2.7 Hz, 1H), 3.80 (d, J = 1.7 Hz, 3H), 3.74 (t, J = 2.6 Hz, 1H), 3.01 (d, J = 12.2 Hz, 1H), 2.43 (qt, J = 13.9, 6.8 Hz, 2H), 2.29 (dd, J = 13.2, 3.4 Hz, 1H), 2.12 (ddd, J = 33.3, 12.2, 6.0 Hz, 2H), 1.98 (dd, J = 15.1, 8.1 Hz, 1H), 1.93 (d, J = 1.8 Hz, 3H), 1.82 (tt, J = 12.8, 2.3 Hz, 2H), 1.76–1.70 (m, 2H), 1.63 (s, 3H), 1.58 (ddd, J = 11.9, 6.9, 2.1 Hz, 1H), 1.54 (t, J = 2.1 Hz, 1H), 1.51 (s, 3H), 1.35 (d, J = 1.8 Hz, 3H), 1.28 (d, J = 14.3 Hz, 1H), 1.25 (d, J = 2.0 Hz, 2H), 1.11 (tdd, J = 19.0, 9.1, 4.3 Hz, 2H), 0.96 (d, J = 1.8 Hz, 3H), 0.90 (s, 6H), 0.90 (dd, J = 18.6, 2.0 Hz, 2H).
^13^C NMR (126 MHz, CDCl_3_) δ 170.6, 170.2, 159.8, 148.2, 132.6, 130.6, 130.4, 128.0, 123.2, 114.1, 77.4, 74.6, 71.5, 68.4, 66.3, 55.4, 49.3, 48.8, 44.0, 39.6, 39.1, 37.2, 36.3, 36.3, 35.7, 32.5, 31.7, 30.4, 30.1, 29.2, 28.4, 25.8, 24.3, 22.9, 21.1, 20.9, 18.0, 17.8, 16.1, 14.3.
HRMS (ESI) m/z calculated for C_39_H_56_O_7_^+^ M + Na^+^: 659.3918; found: 659.3965.
[α]D^26^ = −1.5° (c = 0.68, CH_2_Cl_2_)
Melting point: 193.6 °C
pyridin-2-ylmethyl(Z)-2-(( 3R,4S,5S,8S,9S,10S,11R,13R,14S,16S)-16-acetoxy-3,11-dihydroxy-4,8,10,14-tetramethylhexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoate (2c)
2-Pyridinemethanol (10.5 μL, 0.15 mmol, 1.5 equiv), EDCI (28 mg, 0.15 mmol, 1.5 equiv), and DMAP (12 mg, 0.1 mmol, 1 equiv) were added to a solution of fusidic acid (50 mg, 0.1 mmol, 1.0 equiv) in anhydrous DCM (1 mL). The reaction mixture was stirred at 25 °C under a N_2_ atmosphere. After the reaction was determined to be complete by TLC (24 h), the reaction mixture was diluted with ethyl acetate (10 mL), washed with H_2_O (5 mL), and saturated with aqueous brine (5 mL). The organic layer was then dried over anhydrous MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography (42.5% EA in hexane) on silica gel to afford the product 2c (colorless solid, 36 mg, 60%). R_f_ = 0.4 (60% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 8.57 (d, J = 4.5 Hz, 1H), 7.70 (td, J = 7.7, 1.8 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H), 7.22 (dd, J = 7.6, 4.9 Hz, 1H), 5.88 (d, J = 8.4 Hz, 1H), 5.31 (d, J = 13.5 Hz, 1H), 5.08 (d, J = 13.5 Hz, 2H), 4.34 (q, J = 2.7 Hz, 1H), 3.74 (q, J = 2.8 Hz, 1H), 3.06 (dd, J = 12.8, 3.4 Hz, 1H), 2.49 (m, 2H), 2.31 (dt, J = 13.1, 3.3 Hz, 1H), 2.18 (dd, J = 14.5, 7.4 Hz, 2H), 2.07 (dddd, J = 30.4, 22.9, 12.5, 6.1 Hz, 2H), 1.94 (s, 3H), 1.85 (ddd, J = 15.4, 11.2, 2.8 Hz, 2H), 1.74 (tt, J = 7.7, 5.4 Hz, 2H), 1.63 (d, J = 1.2 Hz, 4H), 1.58 (dq, J = 10.5, 4.0 Hz, 1H), 1.55 (d, J = 2.0 Hz, 1H), 1.54 (d, J = 1.3 Hz, 3H), 1.50 (dq, J = 12.7, 2.7 Hz, 1H), 1.37 (s, 3H), 1.28 (d, J = 14.3 Hz, 1H), 1.26–1.24 (m, 1H), 1.09 (dddd, J = 26.7, 12.4, 8.6, 5.9 Hz, 2H), 0.97 (s, 3H), 0.91 (t, J = 3.4 Hz, 6H).
^13^C NMR (126 MHz, CDCl_3_) δ 170.6, 169.6, 155.7, 149.5, 148.9, 137.5, 132.7, 130.1, 123.2, 122.1, 77.4, 74.5, 71.5, 68.4, 66.6, 49.4, 48.8, 44.2, 39.6, 39.2, 37.2, 36.4, 36.2, 35.7, 32.5, 30.4, 30.1, 29.2, 28.6, 25.8, 24.3, 23.0, 21.1, 20.9, 18.0, 17.9, 16.1.
HRMS (ESI) m/z calculated for C_37_H_53_NO_6_^+^ M + H^+^: 608.3946; found: 608.3979; M + Na^+^: 630.3765; found: 630.3797.
[α]D^26^ = −3.0 (c = 1.64, CH_2_Cl_2_)
Melting point: 115.8 °C
(Z)-2-((4 S,5S,8S,9S,10S,11R,13R,14S,16S)-16-acetoxy-11-hydroxy-4,8,10,14-tetramethyl-3-oxohexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (3a)
(Z)-2-((4 S,5S,8S,9S,10S,13R,14S,16S)-16-acetoxy-4,8,10,14-tetramethyl-3,11-dioxohexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (4a)
Fusidic acid (100 mg, 0.2 mmol, 1 equiv) was dissolved in anhydrous dichloromethane (2.5 mL) while stirring under nitrogen at −10 °C. Dess–Martin periodinane (0.74 mg, 0.17 mmol, 0.9 equiv) was added and the mixture stirred for 3 h. The mixture was diluted with DCM (20 mL) and washed with saturated NaHCO_3_, brine (25 mL each), and dried (Na_2_SO_4_). The organic layer was filtered and reduced in vacuo to leave a clear film. The crude product was then purified by column chromatography (30% (3a), 27.5% (4a), EA in hexane) on silica gel to afford the product 3a (colorless solid, 30 mg, 30%) and 4a (light-yellow solid, 4 mg, 3.7%). R_f_ = 0.5 (3a), 0.6 (4a) (50% EA in hexane):
3a:
^1^H NMR (500 MHz, CDCl_3_) δ 5.92 (d, J = 8.3 Hz, 1H), 5.12 (t, J = 7.1 Hz, 1H),4.42 (m, 1H), 3.07 (d, J = 12.2 Hz 1H), 2.55−2.44 (m, 4H,), 2.43−2.25 (m, 3H), 2.23−2.01 (m, 4H), 1.98 (s, 3H), 2.01−1.86 (m, 4H), 1.68 (s, 3H),1.67−1.64 (m, 1H), 1.62 (s, 3H), 1.35−1.33 (m, 1H), 1.33 (s, 3H), 1.27−1.13 (m, 2H), 1.18 (s, 3H), 1.04 (d, J = 6.6 Hz, 3H), 0.95 (s, 3H);
^13^C NMR (126 MHz, CDCl_3_) δ 213.9, 174.6, 170.7, 151.2, 132.8, 130.0, 123.0, 74.4, 68.2, 48.9, 48.5, 45.8, 45.5, 44.4, 39.5, 39.1, 38.5, 37.0, 36.25, 35.5, 33.7, 28.8, 28.5, 25.8, 24.9, 22.8, 22.0, 20.7, 18.2, 17.9.
4a:
^1^H NMR (500 MHz, CDCl_3_) δ 5.93 (d, J = 8.3 Hz, 1H), 5.08 (t, J = 7.2 Hz, 1H), 2.96–2.86 (m, 2H), 2.78–2.65 (m, 2H), 2.61 (s, 1H), 2.51(ddd, J = 13.7, 8.8, 4.7 Hz, 1H), 2.46–2.32 (m, 3H), 2.27–2.09 (m, 3H), 2.09–1.97 (m, 3H), 2.0 (s, 3H), 1.81 (t, J = 12.1 Hz,1H), 1.67 (s, 3H), 1.60 (s, 3H), 1.60–1.55 (m, 1H), 1.46 (d, J = 14.5 Hz, 1H), 1.26–1.18 (m, 1H), 1.20 (s, 3H), 1.18–1.1 (m, 1H), 1.15 (s, 3H), 1.06–1.02 (m, 6H).
^13^C NMR (126 MHz, CDCl_3_) δ 216.0, 209.6, 174.2, 170.4, 148.2, 133.3, 131.0, 122.6, 74.3, 58.4, 48.8, 47.1, 46.3, 44.9, 44.5, 40.9, 38.1, 36.8, 36.7, 33.4, 32.5, 28.9, 28.1, 25.8, 24.0, 22.6, 21.3, 20.6, 17.9, 17.1, 14.2.
Spectral data for 3a and 4a matched those reported in the literature [50].
(Z)-2-((4S,5S,8S,9S,10S,13R,14S,16S)-16-acetoxy-4,8,10,14-tetramethyl-3,11-dioxohexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (4a)
Fusidic acid (100 mg, 0.2 mmol, 1 equiv) was dissolved in anhydrous dichloromethane (2.5 mL) while stirring under nitrogen at RT. Dess–Martin periodinane (191 mg, 0.45 mmol, 2.25 equiv) was added and the mixture stirred for 15 h. The mixture was diluted with DCM (20 mL) and washed with saturated NaHCO_3_, brine (25 mL each), and dried (Na_2_SO_4_). The organic layer was filtered and reduced in vacuo to leave a clear film. The crude product was then purified by column chromatography 27.5% EA in hexane on silica gel to afford the product 4a (light-yellow solid, 80 mg, 80%). R_f_ = 0.6 (50% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 5.93 (d, J = 8.3 Hz, 1H), 5.08 (t, J = 7.2 Hz, 1H), 2.96–2.86 (m, 2H), 2.78–2.65 (m, 2H), 2.61 (s, 1H), 2.51 (ddd, J= 13.7, 8.8, 4.7 Hz, 1H), 2.46–2.32 (m, 3H), 2.27–2.09 (m, 3H), 2.09–1.97 (m, 3H), 2.0 (s, 3H), 1.81 (t, J= 12.1 Hz,1H), 1.67 (s, 3H), 1.60 (s, 3H), 1.60–1.55 (m, 1H), 1.46 (d, J = 14.5 Hz, 1H), 1.26–1.18 (m, 1H), 1.20 (s, 3H), 1.18–1.1 (m, 1H), 1.15 (s, 3H), 1.06–1.02 (m, 6H).
^13^C NMR (126 MHz, CDCl_3_) δ 216.0, 209.6, 174.2, 170.4, 148.2, 133.3, 131.0, 122.6, 74.3, 58.4, 48.8, 47.1, 46.3, 44.9, 44.5, 40.9, 38.1, 36.8, 36.7, 33.4, 32.5, 28.9, 28.1, 25.8, 24.0, 22.6, 21.3, 20.6, 17.9, 17.1, 14.2.
Spectral data for 4a matched those reported in the literature [50].
(Z)-2-((4 S,5S,8S,9S,10S,11R,13R,14S,16S,E)-16-acetoxy-11-hydroxy-3-(hydroxyimino)-4,8,10,14-tetramethylhexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (3b)
3a (103.0 mg, 0.2 mmol, 1 equiv) was dissolved in isopropanol (1 mL) while stirring under nitrogen, then 50% aqueous hydroxyamine (49 μL 0.8 mmol, 4 equiv) and acetic acid (46 μL, 0.8 mmol, 4 equiv) were added and the mixture warmed to 50 °C and stirred for 15 h. The mixture was reduced in vacuo to leave a clear film. The crude product was then purified by column chromatography (30% EA in DCM) on silica gel to afford the product 3b (colorless solid, 62 mg, 60%). R_f_ = 0.3 (30% EA in DCM):
^1^H NMR (500 MHz, CDCl_3_) δ 5.90 (dd, J = 11.5, 8.4 Hz, 1H), 5.10 (t, J = 7.2 Hz, 1H), 4.37 (s, 1H), 3.26–3.19 (m, 1H), 3.03 (d, J = 12.2 Hz, 1H), 2.68 (q, J = 9.5 Hz, 1H), 2.51–2.43 (m, 2H), 2.33–2.26 (m, 2H), 2.21–2.13 (m, 2H), 2.12–1.95 (m, 2H), 1.94 (d, J = 2.1 Hz, 3H), 1.90 (t, J = 12.3 Hz, 1H), 1.86–1.70 (m, 1H), 1.67 (s, 3H), 1.60 (s, 3H), 1.55 (d, J = 33.4 Hz, 1H), 1.31 (d, J = 23.0 Hz, 3H), 1.39–1.24 (m, 2H), 1.08 (dd, J = 36.3, 6.6 Hz, 3H), 1.07 (s, 2H), 0.93 (d, J = 4.0 Hz, 3H), 0.83 (s, 1H).
^13^C NMR (126 MHz, CDCl_3_) δ 174.9, 174.8, 170.7, 166.2, 163.6, 149.4, 149.0, 132.6, 130.6, 130.4, 123.1, 74.3, 68.1, 67.9, 48.9, 48.7, 48.6, 46.7, 45.2, 44.1, 43.9, 43.6, 39.4, 39.3, 38.9, 37.4, 36.8, 36.6, 36.2, 36.0, 35.4, 34.3, 33.5, 33.1, 32.2, 28.8, 28.3, 25.7, 24.3, 23.9, 22.7, 21.6, 21.2, 20.6, 20.5, 18.3, 17.8, 17.8, 14.2, 14.0.
Spectral data for 3b matched those reported in the literature [40].
(Z)-2-((3 E,4S,5S,8S,9S,10S,11E,13R,14S,16S)-16-acetoxy-3,11-bis(hydroxyimino)-4,8,10,14-tetramethylhexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (4b)
4a (103.0 mg, 0.2 mmol, 1 equiv) was dissolved in isopropanol (1 mL) while stirring under nitrogen. Then, 50% aqueous hydroxyamine (61 μL. 1 mmol, 5 equiv) and acetic acid (58 μL, 1 mmol, 5 equiv) were added and the mixture warmed to 50 °C and stirred for 15 h. The crude was reduced in vacuo to leave a clear film. The crude product was then purified by column chromatography (30% EA in DCM) on silica gel to afford the product 4b (colorless solid, 74 mg, 70%). R_f_ = 0.25 (30% EA in DCM):
^1^H NMR (500 MHz, CDCl_3_) δ 5.65 (d, J = 8.5 Hz, 1H), 4.94 (t, J = 7.4 Hz, 1H), 3.79 (dq, J = 15.5, 5.5 Hz, 1H), 3.22–3.12 (m, 4H), 2.50 (tt, J = 12.8, 5.4 Hz, 1H), 2.34 (dd, J = 13.9, 5.9 Hz, 2H), 2.26–2.20 (m, 1H), 2.14 (td, J = 10.0, 3.3 Hz, 1H), 2.06–1.93 (m, 1H), 1.88 (dd, J = 13.4, 5.4 Hz, 1H), 1.84–1.81 (m, 1H), 1.80 (d, J = 3.5 Hz, 3H), 1.77–1.70 (m, 1H), 1.52 (t, J = 11.6 Hz, 1H), 1.44 (s, 3H), 1.39 (s, 3H), 1.32 (dd, J = 12.0, 5.0 Hz, 1H), 1.13 (dd, J = 14.4, 3.4 Hz, 1H), 1.05 (t, J = 3.3 Hz, 1H), 0.98–0.92 (m, 1H), 0.89 (s, 3H), 0.88 (s, 2H), 0.88 (s, 1H), 0.87 (s, 2H), 0.82 (d, J = 3.2 Hz, 3H), 0.77 (d, J = 3.2 Hz, 3H).
^13^C NMR (126 MHz, CDCl_3_) δ 172.4, 171.5, 167.0, 156.6, 146.1, 132.6, 131.7, 122.9, 74.3, 50.2, 45.0, 44.4, 39.4, 38.1, 36.5, 33.7, 33.2, 32.4, 28.8, 27.9, 25.6, 24.8, 24.3, 23.7, 22.7, 20.4, 20.1, 17.5, 16.6, 14.2.
Spectral data for 4b matched those reported in the literature [40].
(Z)-2-(( 3R,4S,5S,8S,9S,10S,11R,13R,14S,16S)-16-acetoxy-3-(benzyloxy)-11-hydroxy-4,8,10,14-tetramethylhexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (5a)
Fusidic acid (100 mg, 0.2 mmol, 1.0 equiv) was added to a premixed solution of benzoic acid (38 mg, 0.4 mmol, 2.0 equiv), EDCI (75 mg, 0.4 mmol, 2.0 equiv), and DMAP (30 mg, 0.3 mmol, 1.25 equiv) in anhydrous DCM (0.6 mL). The reaction mixture was stirred at 25 °C under a N_2_ atmosphere. After the reaction was determined to be complete by TLC (24 h), the reaction mixture was diluted with ethyl acetate (10 mL), washed with H_2_O (5 mL), and saturated with aqueous brine (5 mL). The organic layer was then dried over anhydrous MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography (25% EA in hexane) on silica gel to afford the product 5a (colorless solid, 29 mg, 24%). R_f_ = 0.5 (50% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 8.05 (dd, J = 8.3, 1.4 Hz, 2H), 7.56 (tt, J = 6.9, 1.4 Hz, 1H), 7.45 (t, J = 7.7 Hz, 2H), 5.91 (d, J = 8.3 Hz, 1H), 5.20 (d, J = 2.7 Hz, 1H), 5.10 (ddd, J = 8.7, 4.5, 1.8 Hz, 1H), 4.36 (q, J = 2.8 Hz, 1H), 3.08 (dd, J = 12.7, 3.2 Hz, 1H), 2.46 (dh, J = 19.2, 6.5 Hz, 2H), 2.36–2.29 (m, 2H), 2.20 (dt, J = 23.9, 7.2 Hz, 3H), 2.12–1.99 (m, 1H), 1.97 (s, 3H), 1.92 (dt, J = 13.0, 3.1 Hz, 2H), 1.89–1.78 (m, 2H), 1.68 (d, J = 8.9 Hz, 2H), 1.67 (s, 3H), 1.59 (d, J = 1.4 Hz, 3H), 1.46 (s, 3H), 1.35 (d, J = 14.4 Hz, 1H), 1.25 (s, 2H), 1.24–1.07 (m, 2H), 1.04 (s, 3H), 0.94 (s, 3H), 0.90 (d, J = 6.8 Hz, 3H).
^13^C NMR (126 MHz, CDCl_3_) δ 174.6, 170.9, 166.3, 150.86, 132.9, 132.7, 131.0, 129.9, 129.6, 128.6, 123.2, 75.1, 74.6, 68.2, 49.1, 48.9, 44.4, 39.6, 39.1, 38.2, 37.1, 35.9, 35.2, 33.0, 31.2, 29.8, 28.8, 28.5, 27.5, 25.8, 25.8, 24.6, 22.7, 20.7, 20.7, 18.2, 17.9, 15.9.
HRMS (ESI) m/z calculated for C_38_H_52_O_7_^+^ M + Na^+^: 643.3605; found: 643.3657.
IR (neat, cm^−1^) 2928, 1713, 1451, 1377, 1271, 1115, 1027, 737, 713.
[α]D^20^ = −20.5 (c = 0.41, CH_2_Cl_2_)
Melting point: 114.2 °C
(Z)-2-(( 3R,4S,8S,9S,10S,11R,13S,14S,16S)-16-acetoxy-11-hydroxy-4,8,10,14-tetramethyl-3-(picolinoyloxy) hexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (5b)
Fusidic acid (100 mg, 0.2 mmol, 1.0 equiv) was added to a premixed solution of picolinic acid (48 mg, 0.4 mmol, 2.0 equiv), EDCI (74 mg, 0.4 mmol, 2.0 equiv), and DMAP (30 mg, 0.2 mmol, 1.25 equiv) in anhydrous DMF (0.6 mL). The reaction mixture was stirred at 25 °C under a N_2_ atmosphere. After the reaction was determined to be complete by TLC (24 h), the reaction mixture was diluted with ethyl acetate (10 mL), washed with H_2_O (5 mL), and saturated with aqueous brine (5 mL). The organic layer was then dried over anhydrous MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography (2% MeOH in DCM) on silica gel to afford the product 5b (colorless solid, 50 mg, 41%). R_f_ = 0.3 (60% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 8.83 (d, J = 5.5 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.89 (td, J = 7.7, 1.8 Hz, 1H), 7.51 (dd, J = 7.7, 4.7 Hz, 1H), 5.93 (d, J = 8.4 Hz, 1H), 5.32 (q, J = 2.9 Hz, 1H), 5.12 (t, J = 7.2 Hz, 1H), 4.39 (q, J = 2.6 Hz, 1H), 3.10 (dd, J = 12.4, 3.2 Hz, 1H), 2.48 (q, J = 8.0 Hz, 2H), 2.35 (td, J = 11.5, 4.3 Hz, 2H), 2.25 (ddt, J = 22.4, 14.7, 6.2 Hz, 2H), 2.20–2.15 (m, 1H), 2.12–2.04 (m, 1H), 2.03–1.99 (m, 1H), 1.97 (s, 3H), 1.95–1.87 (m, 1H), 1.84 (ddd, J = 11.8, 7.2, 3.5 Hz, 2H), 1.68 (s, 3H), 1.62 (d, J = 9.8 Hz, 6H), 1.47 (s, 3H), 1.36 (d, J = 14.2 Hz, 1H), 1.27 (s, 1H), 1.25–1.11 (m, 2H), 1.06 (s, 3H), 0.96 (s, 3H), 0.93 (d, J = 6.7 Hz, 3H).
^13^C NMR (126 MHz, CDCl_3_) δ 174.5, 170.7, 163.7, 150.7, 149.6, 147.7, 138.4, 132.7, 129.9, 127.2, 125.2, 123.2, 77.4, 77.2, 76.9, 76.6, 74.5, 68.2, 49.3, 48.9, 44.4, 39.6, 39.1, 37.9, 37.0, 35.9, 35.4, 32.5, 31.1, 28.9, 28.6, 25.8, 24.4, 23.0, 20.9, 20.7, 18.1, 17.9, 16.0, 15.9.
HRMS (ESI) m/z calculated for C_37_H_51_NO_7_^+^ M + H^+^: 622.3738; found: 622.3778; M + Na^+^: 644.3558; found: 644.3589.
IR (neat, cm^−1^) 2932, 1735, 1438, 1378, 1246, 1141, 1031, 748.
[α]D^20^ = −18.7 (c = 0.3, CH_2_Cl_2_)
Melting point: 130.2 °C
(Z)-2-(( 3R,4S,8S,9S,10S,11R,13S,14S,16S)-16-acetoxy-11-hydroxy-4,8,10,14-tetramethyl-3-(nicotinoyloxy)hexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (5c)
Fusidic acid (100 mg, 0.2 mmol, 1.0 equiv) was added to a premixed solution of nicotinic acid (48 mg, 0.4 mmol, 2.0 equiv), EDCI (74 mg, 0.4 mmol, 2.0 equiv), and DMAP (30 mg, 0.2 mmol, 1.25 equiv) in anhydrous DCM (0.6 mL). The reaction mixture was stirred at 25 °C under a N_2_ atmosphere. After the reaction was determined to be complete by TLC (24 h), the reaction mixture was diluted with ethyl acetate (10 mL), washed with H_2_O (5 mL), and saturated with aqueous brine (5 mL). The organic layer was then dried over anhydrous MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography (25% EA in hexane) on silica gel to afford the product 5c (colorless solid, 68 mg, 56%). R_f_ = 0.3 (60% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 9.32 (s, 1H), 8.89 (s, 1H), 8.49 (d, J = 7.9 Hz, 1H), 7.58 (s, 1H), 5.98 (d, J = 8.3 Hz, 1H), 5.28 (q, J = 2.8 Hz, 1H), 5.11 (t, J = 7.3 Hz, 1H), 4.35 (d, J = 2.8 Hz, 1H), 3.07 (dd, J = 12.5, 3.2 Hz, 1H), 2.47 (t, J = 8.0 Hz, 2H), 2.40 (td, J = 11.7, 4.8 Hz, 1H), 2.36–2.27 (m, 2H), 2.20 (dt, J = 15.4, 7.8 Hz, 2H), 2.14–2.00 (m, 2H), 1.92 (tt, J = 13.2, 6.8 Hz, 3H), 1.86 (s, 3H), 1.83–1.76 (m, 2H), 1.66 (s, 3H), 1.60 (s, 3H), 1.52 (s, 3H), 1.34 (d, J = 14.2 Hz, 1H), 1.25 (q, J = 6.1 Hz, 3H), 1.13 (tt, J = 13.5, 7.3 Hz, 1H), 1.04 (s, 3H), 0.95 (s, 3H), 0.92 (d, J = 6.7 Hz, 3H).
^13^C NMR (126 MHz, CDCl_3_) δ 173.6, 170.5, 164.0, 151.0, 148.9, 148.7, 139.4, 132.4, 130.7, 124.4, 123.3, 123.0, 77.3, 77.2, 77.0, 76.8, 76.5, 74.4, 68.3, 49.2, 48.9, 43.9, 39.5, 39.1, 37.8, 36.9, 35.9, 35.2, 32.3, 31.1, 29.7, 28.9, 28.5, 27.5, 25.8, 24.4, 22.8, 20.7, 20.7, 17.9, 15.8.
HRMS (ESI) m/z calculated for C_37_H_51_NO_7_^+^ M + H^+^: 622.3738; found: 622.3784; M + Na^+^: 644.3558; found: 644.3601.
IR (neat, cm^−1^) 2932, 1720, 1377, 1284, 1125, 1086, 1030, 740, 702.
[α]D^20^ = −16.8 (c = 0.7, CH_2_Cl_2_)
Melting point: 136.2 °C
(Z)-2-(( 3R,4S,5S,8S,9S,10S,11R,13R,14S,16S)-16-acetoxy-11-hydroxy-3-(isonicotinoyloxy)-4,8,10,14-tetramethylhexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (5d)
Fusidic acid (100 mg, 0.2 mmol, 1.0 equiv) was added to a premixed solution of isonicotinic acid (48 mg, 0.4 mmol, 2.0 equiv), EDCI (74 mg, 0.4 mmol, 2.0 equiv), and DMAP (30 mg, 0.2 mmol, 1.25 equiv) in anhydrous DMF (0.6 mL). The reaction mixture was stirred at 25 °C under a N_2_ atmosphere. After the reaction was determined to be complete by TLC (24 h), the reaction mixture was diluted with EA (10 mL), washed with H_2_O (5 mL), and saturated with aqueous brine (5 mL). The organic layer was then dried over anhydrous MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography (1.75% MeOH in DCM) on silica gel to afford the product 5d (colorless solid, 55 mg, 45%). R_f_ = 0.3 (60% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 8.89 (d, J = 5.4 Hz, 2H), 8.09 (d, J = 5.2 Hz, 2H), 5.80 (d, J = 8.3 Hz, 1H), 5.26 (q, J = 2.7 Hz, 1H), 5.14–5.07 (m, 1H), 4.39–4.34 (m, 1H), 3.10 (t, J = 8.6 Hz, 1H), 2.50–2.18 (m, 4H), 2.11 (dq, J = 14.4, 7.6 Hz, 1H), 2.04 (s, 3H), 2.00–1.89 (m, 3H), 1.82 (dtt, J = 19.9, 7.9, 3.9 Hz, 2H), 1.68 (s, 3H), 1.63–1.61 (m, 3H), 1.60 (d, J = 2.0 Hz, 2H), 1.45 (s, 3H), 1.41 (t, J = 7.3 Hz, 1H), 1.35 (d, J = 14.3 Hz, 1H), 1.25 (s, 1H), 1.24–1.11 (m, 2H), 1.05 (s, 3H), 0.95 (s, 3H), 0.90 (d, J = 6.7 Hz, 3H).
^13^C NMR (176 MHz, CDCl_3_) δ 171.0, 164.7, 163.5, 153.0, 132.7, 129.2, 123.0, 74.2, 68.1, 49.0, 48.8, 45.8, 44.6, 44.0, 39.6, 39.6, 39.1, 38.0, 36.9, 36.0, 35.2, 32.4, 30.9, 29.7, 29.1, 29.0, 27.3, 25.8, 24.4, 22.8, 21.2, 20.7, 18.1, 17.9, 15.9, 8.6.
HRMS (ESI) m/z calculated for C_37_H_51_NO_7_^+^ M + Na^+^: 644.3558; found: 644.3603.
IR (neat, cm^−1^) 2961, 2880, 1726, 1377, 1282, 1124, 1031, 483, 418,
[α]D^20^ = −14.4 (c = 0.46, CH_2_Cl_2_)
Melting point: 123.5 °C
(Z)-2-(( 3R,4S,5S,8S,9S,10S,11R,13R,14S,16S)-16-acetoxy-11-hydroxy-4,8,10,14-tetramethyl-3-((pyrazine-2-carbonyl)oxy)hexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (5e)
Fusidic acid (100 mg, 0.2 mmol, 1.0 equiv) was added to a premixed solution of pyrazinoic acid (48 mg, 0.4 mmol, 2.0 equiv), EDCI (74 mg, 0.4 mmol, 2.0 equiv), and DMAP (30 mg, 0.2 mmol, 1.25 equiv) in anhydrous DMF (0.6 mL). The reaction mixture was stirred at 25 °C under a N_2_ atmosphere. After the reaction was determined to be complete by TLC (24 h), the reaction mixture was diluted with ethyl acetate (10 mL), washed with H_2_O (5 mL), and saturated with aqueous brine (5 mL). The organic layer was then dried over anhydrous MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography (3% MeOH in DCM) on silica gel to afford the product 5e (colorless solid, 55 mg, 45%). R_f_ = 0.5 (60% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 9.29 (d, J = 1.4 Hz, 1H), 8.77 (t, J = 1.9 Hz, 1H), 8.76 (d, J = 2.4 Hz, 1H), 5.92 (d, J = 8.4 Hz, 1H), 5.33 (q, J = 2.8 Hz, 1H), 5.10 (t, 1H), 4.36 (q, J = 2.7 Hz, 1H), 3.07 (dd, J = 12.8, 3.2 Hz, 1H), 2.52–2.41 (m, 2H), 2.38–2.26 (m, 2H), 2.26–2.13 (m, 2H), 2.11–2.01 (m, 1H), 1.99 (dd, J = 5.6, 3.0 Hz, 1H), 1.96 (s, 3H), 1.91 (dd, J = 12.8, 2.7 Hz, 1H), 1.89–1.79 (m, 2H), 1.72–1.68 (m, 1H), 1.67–1.66 (m, 5H), 1.65–1.60 (m, 2H), 1.59 (d, J = 1.3 Hz, 3H), 1.46 (s, 3H), 1.35 (d, J = 14.3 Hz, 1H), 1.26–1.09 (m, 2H), 1.05 (s, 3H), 0.94 (s, 3H), 0.93 (d, J = 6.7 Hz, 3H).
^13^C NMR (126 MHz, CDCl_3_) δ 173.8, 170.6, 163.4, 151.0, 147.3, 145.9, 145.0, 144.4, 132.8, 129.7, 123.1, 77.4, 77.1, 74.5, 68.4, 49.2, 49.0, 44.4, 39.7, 39.1, 38.1, 37.1, 36.0, 35.3, 32.7, 31.2, 28.9, 28.5, 27.5, 25.9, 24.5, 22.9, 20.8, 18.2, 17.9, 16.0.
HRMS (ESI) m/z calculated for C_36_H_50_N_2_O_7_^+^ M + Na^+^: 645.3510; found: 645.3554.
IR (neat, cm^−1^) 2929, 2871, 2113, 1732, 1454, 1377, 1342, 1300, 1251, 1237, 1159, 1143, 1052, 1018, 974, 951, 880, 775, 736, 703, 653, 609, 505, 488, 462, 428.
[α]D^20^ = −20.0 (c = 0.27, CH_2_Cl_2_)
Melting point: 118.8 °C
(Z)-2-(( 3R,4S,5S,8S,9S,10S,11R,13R,14S,16S)-16-acetoxy-11-hydroxy-4,8,10,14-tetramethyl-3-((oxazole-4-carbonyl)oxy)hexadecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-6-methylhept-5-enoic acid (5e)
Oxazole-4-carboxylic acid (100 mg, 0.9 mmol, 1.0 equiv) was dissolved in 1 mL of anhydrous tetrahydrofuran (THF). Oxalyl chloride (152 μL, 1.8 mmol, 2.0 equiv) was added dropwise under an inert nitrogen atmosphere, followed by a catalytic amount of DMF (0.1 equiv). The reaction mixture was stirred at room temperature for 4 h. Upon completion, the reaction was monitored by TLC to ensure the formation of the acyl chloride intermediate. The solvent and excess oxalyl chloride were removed under reduced pressure, yielding a yellowish residue. Fusidic acid (153 mg, 0.3 mmol, 0.33 equiv) was dissolved in 1 mL of anhydrous DCM, followed by the addition of pyridine (36 μL, 0.5 mmol, 0.5 equiv) and DMAP (55 mg, 0.5 mmol, 0.5 equiv). The resulting solution was then added dropwise to the previously prepared acyl chloride intermediate under an inert nitrogen atmosphere at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. After the reaction was determined to be complete by TLC (24 h), the reaction mixture was diluted with DCM (10 mL), washed with saturated NaHCO_3_ solution once (5 mL), and water twice (5 mL), and then saturated with aqueous brine (5 mL). The organic layer was then dried over anhydrous MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography (1.5% MeOH in DCM, 2nd column: 30% EA/hexane) on silica gel to afford the product 5e (colorless solid, 42 mg, 35%). R_f_ = 0.5 (60% EA in hexane):
^1^H NMR (500 MHz, CDCl_3_) δ 8.20 (d, J = 1.1 Hz, 1H), 7.95 (d, J = 1.0 Hz, 1H), 5.91 (d, J = 8.4 Hz, 1H), 5.21 (t, J = 2.7 Hz, 1H), 5.10 (t, 1H), 4.36 (d, J = 2.9 Hz, 1H), 3.07 (d, J = 12.3 Hz, 1H), 2.49 (d, J = 7.0 Hz, 1H), 2.46 (d, J = 6.6 Hz, 1H), 2.31 (dt, J = 13.2, 3.3 Hz, 1H), 2.28–2.20 (m, 2H), 2.19 (d, J = 6.5 Hz, 1H), 2.17–2.13 (m, 1H), 2.11–2.03 (m, 1H), 1.99 (d, J = 1.6 Hz, 1H), 1.98 (s, 3H), 1.91 (dd, J = 16.1, 3.5 Hz, 2H), 1.85–1.75 (m, 2H), 1.67 (d, J = 1.4 Hz, 3H), 1.60 (d, J = 1.4 Hz, 4H), 1.59–1.57 (m, 1H), 1.43 (s, 3H), 1.35 (d, J = 14.2 Hz, 1H), 1.26 (d, J = 2.4 Hz, 1H), 1.21–1.16 (m, 1H), 1.11 (dq, J = 13.9, 6.1 Hz, 1H), 1.02 (s, 3H), 0.94 (s, 3H), 0.89 (d, J = 6.7 Hz, 3H).
^13^C NMR (126 MHz, CDCl_3_) δ 173.0, 170.7, 160.6, 151.7, 151.2, 143.6, 133.7, 132.9, 129.5, 123.1, 75.6, 74.5, 68.4, 49.2, 49.0, 44.5, 39.7, 39.1, 38.0, 37.1, 36.0, 35.2, 32.8, 31.2, 28.9, 28.5, 27.5, 25.9, 24.6, 22.9, 20.8, 20.7, 18.2, 17.9, 15.9.
HRMS (ESI) m/z calculated for C_35_H_49_NO_8_^+^ M + Na^+^: 634.3350; found: 634.3394.
IR (neat, cm^−1^) 3462, 3137, 2929, 2873, 1722, 1578, 1517, 1455, 1378, 1253, 1165, 1102, 1056, 1031, 975, 911, 878, 768, 736, 649, 609
[α]D^25^ = −16.7° (c = 0.43, CH_2_Cl_2_)
Melting point: 195.1 °C
3.3. Antibacterial Activity Analysis
Bacterial strains tested were Staphylococcus aureus ATCC 25923 from the American Type Culture Collection (Manassas, VA, USA); E. coli K-12 BW25113 (CGSC #7636); E. coli K-12 JW5503-KanS ΔtolC (CGSC #14206); and E. coli K-12 RFM795 lptD4213 (CGSC #14179) from the Coli Genetics Stock Center (Yale University, New Haven, CT, USA). The ΔtolC strain has a deletion of the entire tolC gene except for nucleotides encoding the last 6 amino acids of this protein and can be considered a null mutant [46]. The lptD4213 mutation has also been called imp-1423 [24]. It consists of a deletion of amino acids 330–352 of the LptD protein. This well-characterized deletion mutant compromises the ability of bacteria to transport LPS into the outer membrane, decreasing the outer membrane permeability barrier.
Minimal inhibitory concentrations (MIC) were determined using Clinical Laboratory Standards Institute (CLSI) reference method equivalent technology as previously described [48]. The fusidic acid stock concentration was 48 mM in DMSO, while the fusidic analogue concentration was 50 mM in DMSO [48]. Compounds were dispensed with a Tecan D300 digital dispenser to create a doubling dilution series for each compound from 128 to 0.03125 μM. Assays were performed in 384-well format, and complete growth inhibition, i.e., the MIC, was scored based on A_600_ as previously described [48]. Values shown represent modal MICs from three technical replicates.
An E. coli S30 extract system for circular DNA (Promega, Madison, WI, USA) was used for coupled in vitro transcription–translation assays set up in 384-well black microplates (PHENIX Research, Swedesboro, NJ, USA). Nanoluciferase (NLuc, 100 ng) expression plasmid and compounds in concentrations ranging from 400 μM to 0.1 μM. in twofold dilutions were added to each reaction, followed by incubation at 37 °C for 60 min. After incubation, PBS containing a 1000-fold dilution of furimazine substrate (Nano-Glo Luciferase Assay System, Promega, Madison, WI, USA) was added to each well, and luminescence was measured using an Infinite M1000 Pro microplate reader (Tecan, Morrisville, NC, USA). Data were collected from three independent experiments. IC_50_ values were calculated from dose–response curves using a three-parameter nonlinear regression model (Prism 10, GraphPad, San Diego, CA, USA).
4. Conclusions
Three classes of fusidic acid analogues were prepared and screened for antibiotic activity. The analogues 2a–c, which probed the effect of carboxylic acid ester functionalization, were inactive against S. aureus and E. coli. The analogues 3a/b and 4a/b, which probed the effect of alcohol oxidation and oxime formation in the A- and C-rings, showed a moderate tolerance for A-ring functionalization, with significant additional reduction in activity with either C-ring functionalization and/or oxime formation. The A-ring ester analogues 5a–f showed a decrease in antibacterial activity against S. aureus and no activity against E. coli. Interestingly the antibacterial activity of the most active ester analogue, 5e, was significantly greater than would be expected based on ribosome inhibitory activity in in vitro translation assays. This suggested that the pyrazine-2-carboxylate ester in the A-ring might spontaneously hydrolyze, conferring on 5e properties of a fusidic acid prodrug. Investigation into the potential of A-ring esters to serve as prodrugs with enhanced pharmacological properties is ongoing and will be reported in due course.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Fernandes P. Fusidic acid: A bacterial elongation factor inhibitor for the oral treatment of acute and chronic staphylococcal infections Cold Spring Harb. Perspect. Med.20166 a 02543710.1101/cshperspect.a 02543726729758 PMC 4691801 · doi ↗ · pubmed ↗
- 2Olaru I.D. Lange C. Indra A. Meidlinger L. Huhulescu S. Rumetshofer R. High Rates of Treatment Success in Pulmonary Multidrug-Resistant Tuberculosis by Individually Tailored Treatment Regimens Ann. Am. Thorac. Soc.2016131271127810.1513/Annals ATS.201512-845OC 27163360 · doi ↗ · pubmed ↗
- 3Cicek-Saydam C. Cavusoglu C. Burhanoglu D. Hilmioglu S. Ozkalay N. Bilgic A. In vitro susceptibility of Mycobacterium tuberculosis to Fusidic acid Clin. Microbiol. Infect.2001770070210.1046/j.1469-0691.2001.00341.x 11843915 · doi ↗ · pubmed ↗
- 4Holmes A.H. Moore L.S. Sundsfjord A. Steinbakk M. Regmi S. Karkey A. Guerin P.J. Piddock L.J. Understanding the mechanisms and drivers of antimicrobial resistance Lancet 201638717618710.1016/S 0140-6736(15)00473-026603922 · doi ↗ · pubmed ↗
- 5Borisova S.A. Guppi S.R. Kim H.J. Wu B. Liu H.-W. O’Doherty G.A. De Novo Synthesis of Glycosylated Methymycin Analogues Org. Lett.2010125150515310.1021/ol 102144 g 20958086 PMC 2980555 · doi ↗ · pubmed ↗
- 6Kim C. Kassu M. Smith K.P. Kirby J.E. Manetsch R. Pyrazole-Thiazole Core-Containing Analogs Exhibit Adjunctive Activity with Meropenem against Carbapenem-Resistant Enterobacteriaceae (CRE)Chem Med Chem 2021162775278010.1002/cmdc.20210032134096189 · doi ↗ · pubmed ↗
- 7Bryskier A. Antimicrobial Agents: Antibacterials and Antifungals 1st ed.ASM Press Washington, DC, USA 200511453
- 8Still J.G. Clark K. Degenhardt T.P. Scott D. Fernandes P. Gutierrez M.J. Pharmacokinetics and safety of single, multiple, and loading doses of Fusidic acid in healthy subjects Clin. Infect. Dis.20115250451210.1093/cid/cir 17421546627 · doi ↗ · pubmed ↗