Preparation and Transformations of Acetophenone-Derived Enamino Ketones, BF2-β-Ketoiminates, and BF2-β-Diketonates
Helena Brodnik, Luka Ciber, Uroš Grošelj, Nejc Petek, Bogdan Štefane, Jurij Svete

TL;DR
This paper reports the synthesis and photochemical transformations of various acetophenone-derived compounds, focusing on the successful De Mayo reaction and novel cyclooctane derivatives.
Contribution
The novel synthesis of diannulated cyclooctane derivatives via photochemical ring-expansion of a BF2-complex.
Findings
Photochemical transformations of BF2-β-diketonate complexes with cycloalkanes yielded De Mayo products in 9–30% yields.
A novel diannulated cyclooctane derivative was obtained from the photochemical ring-expansion of acetyl tetralone-derived BF2-complex 6d.
Attempts to transform enaminones and BF2-β-ketoiminate complexes under similar conditions failed.
Abstract
A series of differently substituted β-enaminones 2a,b, 4a–i, 8a–d, and 9–13, their BF2-β-ketoiminate complexes 5a–d, and BF2-β-diketonate complexes 6a–d were prepared as model substrates for photochemical transformations. The attempted photochemical transformations of enaminones 2, 4, 8 and BF2-β-ketoiminate complexes 5 failed. On the other hand, irradiation of mixtures of BF2-β-diketonate complexes 6a–d and cycloalkanes with UV-A light (365 nm) gave the corresponding De Mayo reaction products 7a–f in 9–30% yields. The photochemical ring-expansion of acetyl tetralone-derived BF2-complex 6d gave novel diannulated cyclooctane derivatives 7e and 7f, which would be difficult to obtain using conventional cyclization methods.
Click any figure to enlarge with its caption.
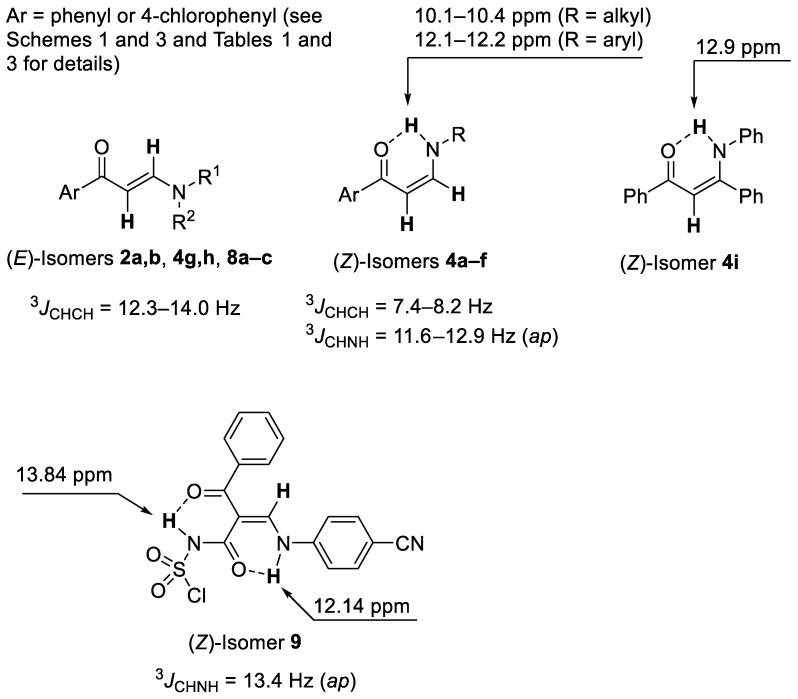 Figure 1
Figure 1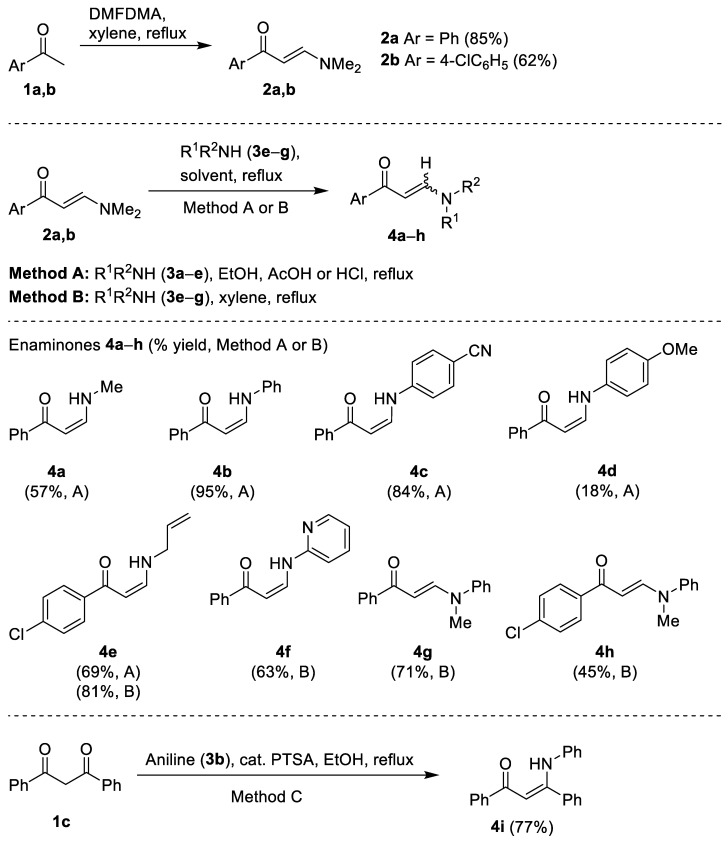 Figure 2
Figure 2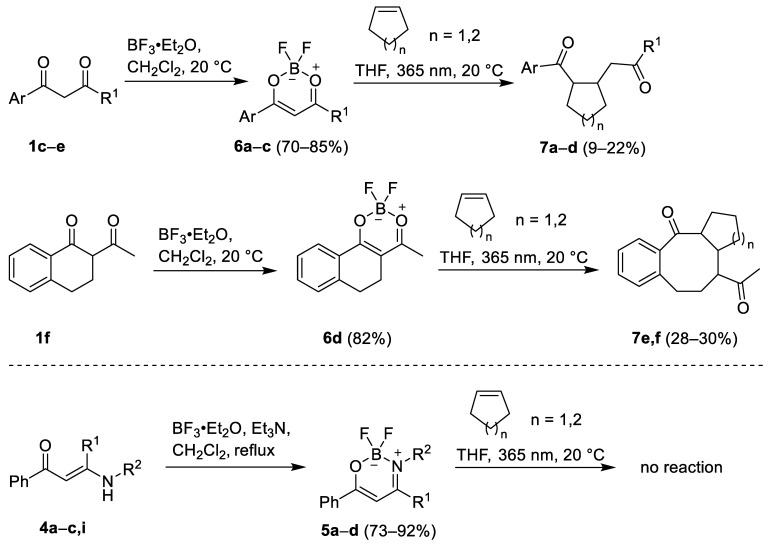 Figure 3
Figure 3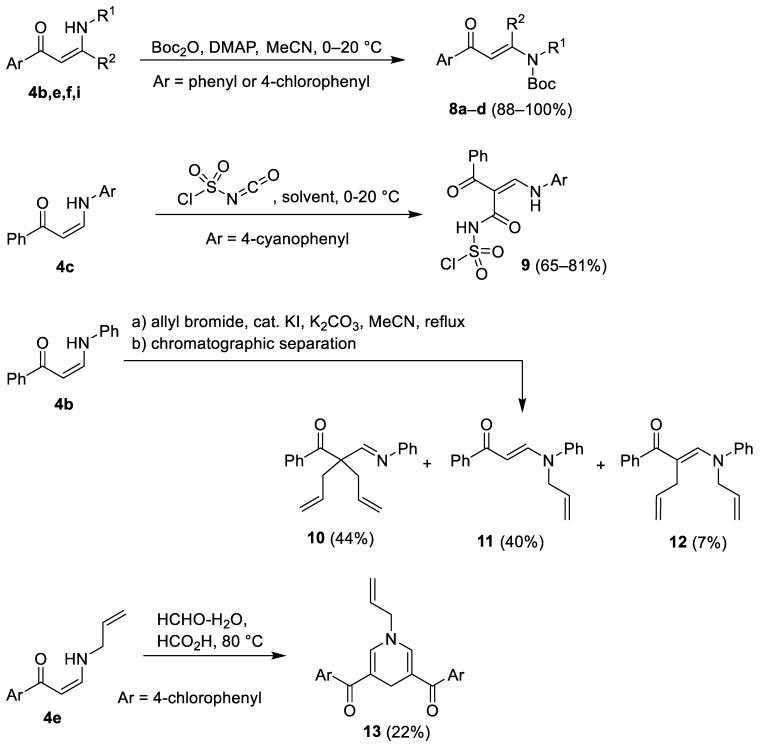 Figure 4
Figure 4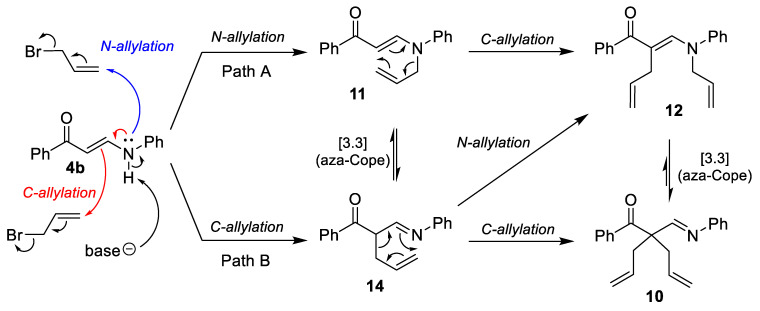 Figure 5
Figure 5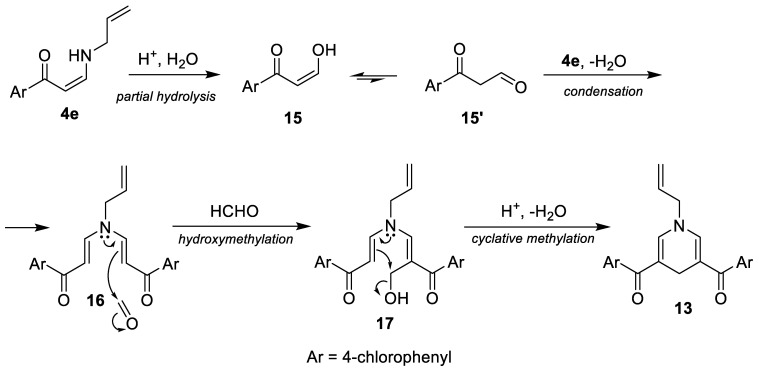 Figure 6
Figure 6- —Slovenian Research and Innovation Agency (ARIS)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and Characterization of Pyrroles · Fluorine in Organic Chemistry · Oxidative Organic Chemistry Reactions
1. Introduction
Enaminones are versatile reagents and building blocks in organic synthesis. Structurally, they are regarded as the enamino equivalents of 1,3-dicarbonyl compounds, as well as the aza-equivalents of enols. Accordingly, these simple and versatile C_3_-building blocks can react as 1,3-dielectrophiles at terminal carbon atoms and/or as nucleophiles at the central carbon atom. Since their synthetic potential was recognized a long time ago, enaminones have a long record of applications, not only in organic and heterocyclic chemistry, but also in other chemistry-related fields and their use in various applications continues. Since the first general review on enaminones by Greenhill in 1977 [1], several reviews on different aspects of chemistry of enaminones have been published [2,3,4,5,6,7,8].
In contrast to well-elaborated thermal transformations, photochemical transformations of enaminones have been less studied. The majority of classical photochemical reactions of enaminones are [2+2] cycloadditions, 6π-electrocyclizations, and ene-type transformations, which have been used in the synthesis of saturated nitrogen heterocycles, indoles, natural products, and their analogues [9,10,11]. Recently, the array of photochemical reactions of enaminones was expanded by photocatalytic transformations, such as α-C–H selenylation, thiocyanation, and perfluoroalkylation. These transformations were also used in the synthesis of 1,2-diketones, β-keto esters, and annulation reactions to obtain five- and six-membered heterocycles [10].
BF_2_-β-diketonates are the cyclic equivalents of 1,3-diketones locked in their enol form. Coordination of β-diketone to Lewis acid (BF_3_·Et_2_O) enhances the electrophilic character of the terminal carbon atoms, while retaining a mild nucleophilic character at the central carbon atom. Similarly, BF_2_-β-ketoiminates are also available from β-enaminones and boron trifluoride etherate. Boron β-diketonates and β-ketoiminates are used as reagents in organic synthesis [12,13,14]. However, they are much better known for interesting optical properties, such as fluorescence, room temperature phosphorescence, and aggregation-induced emission enhancement (AIEE). Therefore, boron β-diketonates and β-ketoiminates are interesting as materials for various optoelectronic applications [15,16,17,18,19,20,21,22,23,24].
In continuation of our long-term interest in the chemistry of enaminones and BF_2_-β-diketonates and their use in organic synthesis, we recently turned our attention to the application of enaminones as ligands and/or building blocks in transition-metal catalysis [25], in asymmetric organocatalysis [26], and in the synthesis of polyenaminones as recyclable optical- and redox-active materials [27]. By extension, we became interested in using enaminones, BF_2_-β-ketoiminates, and BF_2_-β-diketonates as substrates in photochemical and photocatalytic transformations. Therefore, we prepared some model enaminones, BF_2_-β-ketoiminates, and BF_2_-β-diketonates and used them as substrates for further photochemical studies. Herein, we report the results of this study, the synthesis and transformations of selected α-unsubstituted enamino ketones and their BF_2_-complexes, and the photochemical transformations (De Mayo reactions) of BF_2_- β-diketonates.
2. Results
2.1. Synthesis of Model Enamino Ketones 4
(E)-3-(dimethylamino)-1-phenylprop-2-en-1-one (2a) and its 4′-chloro analogue 2b were prepared in two steps from acetophenone (1a) and 4-chloroacetophenone (1b) by heating with 1.1 equiv. of N,N-dimethylformamide dimethylacetal (DMFDMA) in anh. xylene for 16 h to afford the corresponding N,N-dimethylenaminones 2a and 2b in 85% and 62% yield, respectively. Compounds 2a and 2b were then heated with l.1 equiv. of primary amines 3a–e in ethanol in the presence of acetic acid or hydrochloric acid to give the secondary enaminones 4a–e in 18–94% yields (Scheme 1, Method A). On the other hand, acid-catalyzed transamination of N,N-dimethylenaminones 2 did not take place with 2-aminopyridine (3f) and N-methylaniline (3g). Transamination products 4f, 4g, and 4h were then obtained by heating 2a and 2b with excess 3f and 3g in toluene or xylene. In the same way, the secondary enaminone 4e was also obtained in 81% yield from enaminone 2b and excess allylamine (3e) (Scheme 1, Method B). Finally, β-anilinochalcone 4i was prepared by acid-catalyzed condensation of dibenzoylmethane (1c) and aniline (3b) (Scheme 1, Method C). Experimental data for compounds 2 and 4 are summarized in Table 1.
2.2. Synthesis and Photochemical Reactions of BF2-β-Ketoiminates 5 and BF2-β-Diketonates 6
Treatment of secondary enaminones 4a–c,i with BF_3_·Et_2_O in the presence of triethylamine produced the corresponding BF_2_-β-ketoiminate complexes 5a–d in 73–92% yields. Similarly, boron β-diketonates 6a–d were prepared from 1,3-diketones 1c–f and BF_3_·Et_2_O in dichloromethane following the literature procedure [28]. BF_2_-β-diketonate complexes 6 reacted readily with cyclopentene and cyclohexene under irradiation (365 nm) in dichloromethane to furnish the corresponding cycloalkane insertion products 7a–f in 9–30% yields. Gratifyingly, novel compounds 7e and 7f are nice examples of a ring-expansion reaction furnishing cyclooctane ring, which is difficult to obtain using conventional cyclization methods (Scheme 2). On the other hand, attempts to carry out De Mayo reaction by irradiation of mixtures of β-ketoiminate complexes 5a–d and various alkenes with UV-A light (365 nm) failed. This is in line with, to our knowledge, the only literature report on De Mayo reaction using BF_2_-β-ketoiminates as substrates [29]. Experimental data for compounds 5–7 are summarized in Table 2.
2.3. Other Transformations of Model Enamino Ketones 4
As the initially prepared substrates 4 and 5 failed to produce the desired cycloalkane insertion products, additional derivatives of secondary enaminones were prepared and tested for photochemical conversion. Compounds 4b,e,f,i were acylated with di-tert-butyl dicarbonate (Boc_2_O) in acetonitrile in the presence of 4-(dimethylamino)pyridine (DMAP) to afford the corresponding N-Boc derivatives 8a–d in 88–100% yields. On the other hand, reaction of 4c with chlorosulfonyl isocyanate (CSI) in Et_2_O at 0–20 °C furnished (Z)-{2-benzoyl-3-[(4-cyanophenyl)amino]acryloyl}sulfamoyl chloride (9) as the C-acylation product in 80% yield. Replacement of Et_2_O with toluene and acetonitrile as solvents gave 9 in 81% and 65% yield, respectively. Somewhat surprisingly, treatment of 4b with excess allyl bromide in the presence of K_2_CO_3_ gave a mixture of three products 10–12. Subsequent chromatographic separation afforded the C,C-diallylation product 10 in 44% yield, the N-monoallylation product 11 in 40% yield, and the C,N-diallylation product 12 in 7% yield. Attempted methylation of 4e with aq. formaldehyde in formic acid did not give any of the expected methylation products. Instead, 1-allyl-3,5-bis(4-chlorobenzoyl)-1,4-dihydropyridine (13) was isolated in 22% yield (Scheme 3). Experimental data for compounds 8–13 are summarized in Table 3.
Formations of the products 4–9 (cf. Scheme 1, Scheme 2 and Scheme 3) were expected and are explainable by general mechanisms for enaminone transamination (compounds 4), boron β-ketoiminate and β-diketonate complex formation (compounds 5 and 6), De Mayo reaction (compounds 7), acylation (compounds 8), and electrophilic substitution (compound 9). On the other hand, formation of a mixture of three allylation products 10–12 and dihydropyridine derivative 13 were outside the expected product formation and require some further discussion. The proposed reaction pathway leading to a mixture of 10–12 is shown on Scheme 4. Reaction of enaminone 4b with 2 equiv. allyl bromide can take place as initial N-allylation (Path A) and/or C-allylation (Path B) to give the respective monoallylated compounds 11 and/or 14. Further allylation of the N-allyl intermediate 11 can only take place at the carbon atom to give the C,N-diallylated compound 10, while allylation of the C-allyl intermediate 14 can give two products, the C,C-diallylated product 10 and the C,N-diallylated compound 12. Finally, the allyl group in monoallylated compounds 11 and 14 and in diallylated compounds 10 and 12 can migrate from carbon to nitrogen and vice versa via a [3.3] sigmatropic rearrangement (aza-Cope rearrangement) [30,31,32] as proposed in Scheme 4.
A plausible explanation for the formation of 1,4-dihydropyridine derivative 13 is shown in Scheme 5. Under aqueous acidic conditions, the enamine moiety of N-allylenaminone 4e partially hydrolyzes to give the β-keto aldehyde intermediate 15/15′, which reacts with enaminone 4e to afford the bis-enaminone intermediate 16. Subsequent C-hydroxymethylation of 16 with formaldehyde gives the intermediate 17, which undergoes acid-catalyzed cyclative alkylation via elimination of water to furnish the 1,4-dihydropyridine derivative 13.
Unfortunately, the attempted photochemical and photocatalytic transformations of compounds 4 and 8 were unsuccessful. In most cases, no reaction took place, while in some cases, complex inseparable mixtures of products were obtained.
3. Structure Determination and Compound Characterization
The structures of compounds 4–13 were determined by spectroscopic methods (^1^H, ^13^C NMR, IR, HRMS, and UV-vis) and by elemental analyses for C, H, and N. The structures of compounds 5b, 8a, 8c, and 8d (Figures S2–S5) in the solid state were determined by X-Ray diffraction.
In solution, enaminones can exist as mixtures of the E- and the Z-isomers and the predominance of either isomer depends on the structure of enaminone and the solvent [1,2,3,4,5,6,7,8]. NMR spectra of tertiary enaminones 2a,b, 4g,h, and 8a–c in CDCl_3_ exhibited a large vicinal coupling constant, ^3^JCHCH = 12.3–14.0 Hz, which was in agreement with the E-configuration around the C=C double bond. On the other hand, the NMR spectra of secondary enaminones 4a–f in CDCl_3_ exhibited smaller vicinal coupling constant, ^3^JCHCH = 7.4–8.2 Hz, which was in agreement with the Z-configuration around the C=C double bond. The Z-configuration of 4a–f in CDCl_3_ is favored by the intramolecular N–H···O=C hydrogen bond, which is consistent with the observed chemical shift, δ = 10.1–12.2 ppm, for the NH proton. On the basis of the large δ chemical shift around 12.5 ppm, the Z-configuration around the C=C double bond was also assigned to compounds 4i and 9. Furthermore, the chemical shift of the sulfonamide NH proton, δ = 13.9 ppm, supports the intramolecular N–H···O=C hydrogen bond between the sulfonamide and the benzoyl groups attached to the α-carbon atom of 9 (Figure 1).
The absorption spectra of compounds 4, 5, 8, and 9 are shown in Figure S6. Absorption spectra of N-aryl-substituted enaminones 4b–d,f,g,i show typical absorption maxima between 350 nm (4g, purple line) and 380 nm (4d, blue line) (Figure S6A). Absorption spectra of oxazaborinines 5 are similar to those of parent enaminones 4; the N-methyloxazaborinine 5a absorbs at 334 nm (brown line), while the N-aryl analogues 5b–d absorb between 356 nm (5d, green line) and 373 nm (5c, blue line) (Figure S6B). N-acylation of 4 into product 8 resulted in the blue-shift of absorption maxima, which appear between 300 nm (8c, red line) and 327 nm (8d, blue line) (Figure S6C). Compound 9 with chlorosulfonylaminocarbonyl group at α-position exhibits two distinct absorption maxima at 267 nm and 360 nm (Figure S6D). In summary, the N-aryl-substituted enaminones 4b–d,f,g,i and 9 and the respective BF_2_-complexes 5b–d absorb visible light up to 415–470 nm, while the N-Boc-analogues 8a,c,d and N-methyloxazaborinine 5a absorb only UV-A light below 400 nm (Figure S6).
4. Experimental
4.1. General Methods
Melting points were determined on a Kofler micro hot stage and on a Mettler Toledo MP30 automated melting point system (Mettler Toledo, Columbus, OH, USA). The NMR spectra were recorded in CDCl_3_ and DMSO-d_6_ using Me_4_Si as the internal standard on a Bruker Avance III Ultrashield 500 and Bruker Avance Neo 600 instruments (Bruker, Billerica, MA, USA) at 500 and 600 MHz for ^1^H and at 125 and 150 MHz for ^13^C nucleus, respectively. Chemical shifts (δ) are given in ppm relative to Me_4_Si as internal standard (δ = 0 ppm) and vicinal coupling constants (J) are given in hertz (Hz). HRMS spectra were recorded on an Agilent 6224 time-of-flight (TOF) mass spectrometer equipped with a double orthogonal electrospray source under atmospheric pressure ionization (ESI) coupled to an Agilent 1260 high-performance liquid chromatograph (HPLC) (Agilent Technologies, Santa Clara, CA, USA). UV-vis spectra were recorded in MeOH using a Varian Cary Bio50 UV–Visible Spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Emission spectra were recorded on a PerkinElmer LS 50 B Luminescence spectrophotometer (PerkinElmer, Waltham, MA, USA). Fourier-transform infrared (FT-IR) spectra were obtained on a Bruker FTIR Alpha Platinum spectrophotometer (Bruker, Billerica, MA, USA) using attenuated total reflection (ATR) sampling technique. Microanalyses for C, H, and N were obtained on a Perkin-Elmer CHNS/O Analyzer 2400 Series II (PerkinElmer, Waltham, MA, USA). Column chromatography (CC) was performed on silica gel (Silica gel 60, particle size: 0.035–0.070 mm (Sigma-Aldrich, St. Louis, MO, USA)). Photochemical transformations were performed on a Penn PhD Photoreactor M2 with LED light source (3 W, 365 nm) and air-cooling (Penn Photon Devices, Pennsburg, PA, USA).
Acetophenones 1a and 1b, 1,3-diketones 1c–f, N,N-dimethylformamide dimethylacetal (DMFDMA), amines 3a–g, acetic acid, 37% hydrochloric acid, di-tert-butyl dicarbonate (Boc_2_O), chlorosulfonyl isocyanate (CSI), allyl bromide, 37% aqueous formaldehyde, formic acid, cyclopentene, cyclohexene, triethylamine, and BF_3_·Et_2_O are commercially available (Sigma-Aldrich, St. Louis, MO, USA).
4.2. General Procedure for the Synthesis of N,N-Dimethylenaminones 2a and 2b
N,N-Dimethylformamide dimethyl acetal (DMFDMA) (1.5 mL, 11.2 mmol) was added to a solution of acetophenone derivative 1 (1.20 g, 10 mmol) in anh. xylene (10 mL) and the mixture was refluxed under argon for 16 h. Volatile components were evaporated in vacuo and the orange–red solid residue was triturated with petroleum ether (20 mL). The precipitate was collected by filtration, washed with petroleum ether (2 × 5 mL), and air-dried to give 2.
4.2.1. (E)-3-(Dimethylamino)-1-phenylprop-2-en-1-one (2a)
Prepared from acetophenone (1a) (1.20 g, 10 mmol) and DMFDMA (1.5 mL, 11.2 mmol). Yield: 1.49 g (85%) of ochre solid, m.p. 93–94 °C, lit. [33] m.p. 90–92 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 7.89 (dt, J = 6.9, 1.5 Hz, 2H), 7.80 (d, J = 12.4 Hz, 1H), 7.48–7.36 (m, 3H), 5.71 (d, J = 12.4 Hz, 1H), 3.14 and 2.92 (2 br s, 1:1, 6H). ^13^C NMR (126 MHz, CDCl_3_) δ 188.6, 154.2, 140.5, 130.8, 128.1, 127.5, 92.2, 45.0, 37.2. νmax 2905, 2805, 1633, 1581, 1428, 1362, 1273, 1051 cm^−1^; ESI-HRMS: m/z = 176.1068 [M+H]^+^, C_11_H_14_NO requires m/z = 176.1070. Anal. Calcd. for C_11_H_13_NO: C, 75.40; H, 4.93; N, 7,90%. Found: C, 75.59; H, 4.93, N, 7.90%. Physical and spectral data are in agreement with the literature data [33,34,35].
4.2.2. (E)-1-(4-Chlorophenyl)-3-(dimethylamino)prop-2-en-1-one (2b)
Prepared from 4-chloroacetophenone (1b) (1.55 g, 10 mmol) and DMFDMA (1.5 mL, 11.2 mmol). Yield: 1.295 g (62%) of light orange solid, m.p. 87–88 °C, lit. [34] m.p. 85–87 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 7.87–7.81 (m, 2H), 7.80 (d, J = 12.3 Hz, 1H), 7.40–7.34 (m, 2H), 5.66 (d, J = 12.3 Hz, 1H), 3.16 and 2.93 (2 br s, 1:1, 6H). Physical and spectral data are in agreement with the literature data [34].
4.3. General Procedures for the Synthesis of Enaminones 4a–h
General Procedure A (G.P.A).
A mixture of N,N-dimethylenaminone 2 (10 mmol), amine 3 (10 mmol), ethanol (40 mL), and acetic acid (1.5 mL, 25 mmol) or 37% hydrochloric acid (1 mL, 10 mmol), was stirred under reflux for 3–16 h. The reaction mixture was cooled to 0 °C (ice bath), the precipitate was collected by filtration and washed with EtOH–water (1:1, 2 × 5 mL) to give compound 4.
General Procedure B (G.P.B).
A two-necked 50 mL flask was charged with N,N-dimethylenaminone 2 (5 mmol), anh. xylene (25 mL), and amine 3 (5 mmol). The flask was then equipped with a reflux condenser and nitrogen inlet adapter. The reaction mixture was stirred under reflux and dry nitrogen was slowly bubbled into the reaction mixture to assist the removal of dimethylamine, evolution of which was monitored by indication with wet litmus paper at the top of reflux condenser. After approximately 12 h, the evolution of dimethylamine ceased (negative wet-litmus test) indicating completion of transamination reaction. Volatile components were evaporated in vacuo and the residue was purified by CC. Fractions containing the product were combined and evaporated in vacuo to give compound 4.
4.3.1. (Z)-3-(methylamino)-1-phenylprop-2-en-1-one (4a)
Prepared from enaminone 2a (1.754 g, 10 mmol) and methylamine hydrochloride (3a) (1.35 g, 20 mmol), G.P.A, reflux for 16 h. Addition of acetic acid or hydrochloric acid was omitted and the product 4a did not precipitate for the reaction mixture. Volatile components were evaporated in vacuo and the residue was purified by CC (EtOAc–hexanes, 1:5). Fractions containing the product were combined and evaporated in vacuo to give compound 4a. Yield: 912 mg (57%) of yellow solid, m.p. 135–137 °C, lit. [36] m.p. 138–139 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 10.13 (s, 1H), 7.83–7.74 (m, 2H), 7.45–7.27 (m, 3H), 6.83 (dd, J = 12.9, 7.4 Hz, 1H), 5.61 (d, J = 7.3 Hz, 1H), 2.99 (d, J = 5.1 Hz, 3H). νmax 3071, 2948, 1630 (C=O), 1512, 1422, 1395, 1286, 1058 cm^−1^. Physical and spectral data are in agreement with the literature data [36,37].
4.3.2. (Z)-1-Phenyl-3-(phenylamino)prop-2-en-1-one (4b)
Prepared from enaminone 2a (1.754 g, 10 mmol) and aniline hydrochloride (3b) (1.30 g, 10 mmol), G.P.A, reflux for 3 h. Yield: 2.111 g (95%) of yellow solid, m.p. 139–140 °C, lit. [38] m.p. 141 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 12.15 (d, J = 12.3 Hz, 1H), 7.98–7.92 (m, 2H), 7.57–7.42 (m, 4H), 7.39–7.32 (m, 2H), 7.15–7.06 (m, 3H), 6.04 (d, J = 7.8 Hz, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 191.0, 145.0, 140.2, 139.2, 131.6, 129.8, 128.5, 127.3, 123.7, 116.4, 93.7. νmax 3050, 1659 (C=O), 1542, 1472, 1268, 1239, 1175, 886 cm^−1^. ESI-HRMS: m/z = 224.1082 [M+H]^+^, C_15_H_14_NO requires m/z = 224.1070. Physical and spectral data are in agreement with the literature data [38,39].
4.3.3. (Z)-4-[(3-Oxo-3-phenylprop-1-en-1-yl)amino]benzonitrile (4c)
Prepared from enaminone 2a (526 mg, 3 mmol), 4-aminobenzonitrile (3c) (390 mg, 3.3 mmol), and acetic acid (1.5 mL), G.P.A, reflux for 16 h. Yield: 622 mg (84%) of yellow solid, m.p. 153–154 °C, lit. [40] m.p. of the (E)-isomer 209–210 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 12.18 (d, J = 11.9 Hz, 1H), 7.98–7.91 (m, 2H), 7.66–7.59 (m, 2H), 7.58–7.50 (m, 1H), 7.53–7.43 (m, 3H), 7.17–7.08 (m, 2H), 6.17 (d, J = 8.1 Hz, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 191.9, 144.0, 142.6, 138.5, 134.0, 132.3, 128.6, 127.5, 118.9, 116.0, 106.0, 96.3. νmax 3056, 2223 (C≡N), 1643 (C=O), 1471, 1287, 1243, 1176, 1019 cm^−1^; ESI-HRMS: m/z = 249.1015 [M+H]^+^, C_16_H_13_N_2_O requires m/z = 249.1022. Anal. Calcd. for C_16_H_12_N_2_O: C, 77.40; H, 4.87; N, 11.28%. Found: C, 77.04; H, 4.93, N, 10.90%. Physical and spectral data are in agreement with the literature data [40].
4.3.4. (Z)-3-[(4-Methoxyphenyl)amino]-1-phenylprop-2-en-1-one (4d)
Prepared from enaminone 2a (175 mg, 1 mmol), 4-methoxyaniline (3d) (123 mg, 1 mmol), and acetic acid (0.5 mL), G.P.A, reflux for 16 h. The product was purified by CC (Et_2_O). Fractions containing the product were combined and evaporated in vacuo to give compound 4d. Yield: 45 mg (18%) of yellow solid, m.p. 146–148 °C, lit. [41] 145–147 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 12.20 (d, J = 12.4 Hz, 1H), 7.97–7.90 (m, 2H), 7.53–7.40 (m, 4H), 7.09–7.02 (m, 2H), 6.93–6.86 (m, 2H), 5.98 (d, J = 7.7 Hz, 1H), 3.80 (s, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 190.6, 156.4, 145.9, 139.3, 133.8, 131.4, 128.4, 127.2, 117.9, 115.0, 92.9, 55.6. νmax 3060, 2990, 1624 (C=O), 1470, 1290, 1254, 1179, 1030 cm^−1^. ESI-HRMS: m/z = 254.1176 [M+H]^+^, C_16_H_16_NO_2_ requires m/z = 254.1176. Physical and spectral data are in agreement with the literature data [41].
4.3.5. (Z)-3-(Allylamino)-1-(4-chlorophenyl)prop-2-en-1-one (4e)
Prepared from enaminone 2b (210 mg, 1 mmol), allylamine (3e) (90 μL, 1.2 mmol), and 37% aq. HCl (3 drops, 1 mmol), reflux for 16 h, G.P.A. The product 4e did not precipitate for the reaction mixture. Volatile components were evaporated in vacuo and the residue was purified by CC (EtOAc–hexanes, 1:1). Fractions containing the product were combined and evaporated in vacuo to give compound 4e. Prepared also from enaminone 2b (2.10 g, 10 mmol) and allylamine (3e) (1.8 mL, 24 mmol) in anh. xylene (25 mL), G.P.B, CC (EtOAc–hexanes, 1:1). Yield: 152 mg (69%, G.P.A) and 1.796 g (81%, G.P.B) of brownish solid, m.p. 60–61 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 10.35 (s, 1H), 7.81 (d, J = 8.6 Hz, 2H), 7.38 (d, J = 8.6 Hz, 2H), 6.95 (dd, J = 12.8, 7.5 Hz, 1H), 5.90 (ddt, J = 17.1, 10.4, 5.4 Hz, 1H), 5.69 (d, J = 7.4 Hz, 1H), 5.28 (dq, J = 17.2, 1.6 Hz, 1H), 5.22 (dq, J = 10.2, 1.4 Hz, 1H), 3.89 (tt, J = 5.7, 1.7 Hz, 2H). ^13^C NMR (126 MHz, CDCl_3_) δ 188.6, 154.4, 138.1, 137.0, 134.1, 128.5, 128.5, 117.3, 90.4, 51.1. νmax 3278, 2909, 1533, 1274, 1087, 1006, 935, 847, 762 cm^−1^. ESI-HRMS: m/z = 222.0608 [M+H]^+^, C_12_H_13_ClNO requires m/z = 222.0677.
4.3.6. (Z)-1-Phenyl-3-[(pyridin-2-yl)amino]prop-2-en-1-one (4f)
Prepared from enaminone 2a (876 mg, 5 mmol) and 2-aminopyridine (3f) (470 mg, 5 mmol) in anh. xylene (25 mL), G.P.B, CC (Et_2_O). Yield: 710 mg (63%) of yellow solid, m.p. 126–128 °C, lit. [42] m.p. 169–170 °C, lit. [43] m.p. 93 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 12.16 (d, J = 11.6 Hz, 1H), 8.31 (ddd, J = 4.9, 1.9, 0.8 Hz, 1H), 8.26 (dd, J = 11.6, 8.2 Hz, 1H), 8.00–7.93 (m, 2H), 7.62 (ddd, J = 8.1, 7.3, 1.9 Hz, 1H), 7.56–7.48 (m, 1H), 7.50–7.43 (m, 2H), 6.95 (ddd, J = 7.3, 4.9, 0.9 Hz, 1H), 6.83 (dt, J = 8.2, 0.9 Hz, 1H), 6.15 (d, J = 8.2 Hz, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 191.9, 151.7, 148.5, 142.9, 139.0, 138.4, 131.8, 128.5, 127.5, 118.5, 111.7, 95.2. νmax 3061, 1591, 1536, 1470, 1416, 1230, 1202, 1016 cm^−1^. ESI-HRMS: m/z = 225.1018 [M+H]^+^, C_14_H_13_N_2_O requires m/z = 225.1022. Physical and spectral data are in agreement with the literature data [42,43].
4.3.7. (E)-3-[Methyl(phenyl)amino]-1-phenylprop-2-en-1-one (4g)
Prepared from enaminone 2a (876 mg, 5 mmol) and N-methylaniline (3g) (470 mg, 5 mmol) in anh. xylene (25 mL), G.P.B, CC (Et_2_O). Yield: 840 mg (71%) of beige solid, m.p. 99–101°C, lit. [44] m.p. 110–112 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.23 (d, J = 12.7 Hz, 1H), 7.98–7.91 (m, 2H), 7.53–7.46 (m, 1H), 7.48–7.41 (m, 2H), 7.42–7.34 (m, 2H), 7.25–7.14 (m, 3H), 6.10 (d, J = 12.7 Hz, 1H), 3.40 (s, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 189.41, 149.95, 146.4, 140.0, 131.4, 129.5, 128.3, 127.7, 124.9, 112.4, 96.9, 37.4. νmax 3052, 3034, 1650 (C=O), 1539, 1491, 1260, 1176, 1020 cm^−1^. ESI-HRMS: m/z = 238.1222 [M+H]^+^, C_16_H_16_NO requires m/z = 238.1226. Anal. Calcd. for C_16_H_15_NO: C, 80.98; H, 6.37; N, 5.90%. Found: C, 80.59; H, 6.51, N, 5.98%. Physical and spectral data are in agreement with the literature data [44].
4.3.8. (E)-1-(4-Chlorophenyl)-3-[methyl(phenyl)amino]prop-2-en-1-one (4h)
Prepared from enaminone 2b (323 mg, 1.5 mmol) and N-methylaniline (3g) (400 μL, 3.7 mmol) in anh. xylene (5 mL), G.P.B, CC (Et_2_O). Yield: 183 mg (45%) of yellow solid, m.p. 119–120 °C, lit. [45] m.p. 119–120 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.22 (d, J = 12.6 Hz, 1H), 7.89 (d, J = 8.5 Hz, 2H), 7.43–7.37 (m, 4H), 7.23–7.17 (m, 3H), 6.04 (d, J = 12.6 Hz, 1H), 3.41 (s, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 187.9, 150.4, 146.3, 138.3, 137.5, 129.6, 129.1, 128.5, 125.2, 120.6, 96.3, 37.2. νmax 3065, 2916, 2198, 2163, 2051, 1923, 1675 (C=O), 1639 (C=O), 1572, 1539, 1497, 1466, 1426, 1392, 1347, 1326, 1305, 1267, 1208, 1182, 1158, 1137, 1087, 1056, 1030, 1009, 973, 901, 874, 845, 800, 758, 742, 713, 695, 679, 626, 610 cm^−1^. ESI-HRMS: m/z = 272.0835 [M+H]^+^, C_16_H_15_ClNO requires m/z = 272.0837. Physical and spectral data are in agreement with the literature data [45].
4.4. (Z)-1,3-Diphenyl-3-(phenylamino)prop-2-en-1-one (4i)
This compound was prepared by a modified literature procedure [46]. Dibenzoylmethane (12b) (11.2 g, 50 mmol) and para-toluenesulfonic acid monohydrate (476 mg, 2.5 mmol) were added to a solution of aniline (3b) (4.56 mL, 5 mmol) in ethanol (100 mL) and the mixture was stirred under reflux for 16 h. Water (50 mL) and charcoal (~1 g) were added, the mixture was stirred under reflux for 5 min, the suspension filtered hot, and the filtrate was left to stand at ~4 °C for 12 h. The precipitate was collected by filtration and re-crystallized from EtOH–H_2_O (2:1, 250 mL) to give pure compound 4i. Yield: 11.50 g (77%) of yellow crystals, m.p. 102–103 °C, lit. [46] m.p. 102–103 °C, lit. [47] m.p. 144–145 °C, lit. [48] m.p. 101–102 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 12.90 (s, 1H), 8.00–7.94 (m, 2H), 7.53–7.30 (m, 8H), 7.17–7.09 (m, 2H), 7.03–6.95 (m, 1H), 6.82–6.77 (m, 2H), 6.09 (s, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ ^13^C NMR (126 MHz, CDCl_3_) δ 189.8, 161.6, 140.0, 139.6, 136.0, 131.4, 129.8, 128.9, 128.7, 128.5, 127.4, 124.3, 123.4, 97.2. νmax 3019, 1591, 1555, 1510, 1319, 1284, 1215, 1053 cm^−1^. ESI-HRMS: m/z = 300.1380 [M+H]^+^, C_21_H_18_NO requires m/z = 300.1383. Physical and spectral data are in agreement with the literature data [46,47,48].
4.5. General Procedure for the Synthesis of β-Ketoiminate Complexes 5a–d
Compounds 5 a–d were prepared by a modified literature procedure [13,14]. Under argon, Et_3_N (140 µL, 1 mmol) and BF_3_·Et_2_O (370 µL, 3 mmol) were added subsequently to a stirred suspension of enaminone 4 (1 mmol) in anh. dichloromethane (5 mL) and the mixture was stirred under reflux under argon for 4–24 h.
Workup A: Volatile components were evaporated in vacuo and the residue was purified by CC (dry load). Fractions containing the product were combined and evaporated in vacuo to give 5.
Workup B: Volatile components were evaporated in vacuo and the residue was triturated with Et_2_O (5 mL) and water (5 mL). The precipitate was collected by filtration and washed with Et_2_O (5 mL) to give 5.
4.5.1. 2,2-Difluoro-3-methyl-6-phenyl-2H-1,3λ4,2λ4-oxazaborinine (5a)
Prepared from enaminone 4a (162 mg, 1 mmol), Et_3_N (140 µL, 1 mmol), and BF_3_·Et_2_O (370 µL, 3 mmol), reflux for 24 h, Workup A, CC (CH_2_Cl_2_). Yield: 164 mg (78%) of beige solid, m.p. 128–129 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 7.96–7.89 (m, 2H), 7.69 (p, J = 4.5 Hz, 1H), 7.56–7.48 (m, 1H), 7.45 (dd, J = 8.4, 6.9 Hz, 2H), 6.09 (d, J = 5.4 Hz, 1H), 3.40 (s, 3H). ^13^C NMR (126 MHz, CDCl_3_) δ 172.1, 161.7, 133.1, 132.6, 128.8, 127.5, 91.9, 39.6. νmax 2921, 1643, 1551, 1491, 1380, 1151, 1013, 893 cm^−1^. ESI-HRMS: m/z = 209.0936 [M]^+^, C_10_H_10_BF_2_NO requires m/z = 209.0933. Anal. Calcd. for C_10_H_10_BF_2_NO: C, 57.47; H, 4.82; N, 6.70%. Found: C, 57.78; H, 4.67, N, 6.60%.
4.5.2. 2,2-Difluoro-3,6-diphenyl-2H-1,3λ4,2λ4-oxazaborinine (5b)
Prepared from enaminone 4b (894 mg, 4 mmol), Et_3_N (560 µL, 4 mmol) and BF_3_·Et_2_O (1.48 mL, 12 mmol), reflux for 16 h, Workup B. Yield: 797 mg (73%) of bright yellow solid, m.p. 194–195 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.06–8.00 (m, 2H), 7.95 (s, 1H), 7.61–7.55 (m, 1H), 7.52–7.43 (m, 6H), 7.41–7.36 (m, 1H), 6.36 (d, J = 5.8 Hz, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 174.3, 159.6, 142.9, 133.3, 132.7, 129.7, 129.0, 128.3, 128.0, 123.5, 93.7. νmax 1609, 1534, 1487, 1364, 1120, 1082, 1015, 998 cm^−1^. ESI-HRMS: m/z = 271.1090 [M+H]^+^, C_15_H_13_BF_2_NO requires m/z = 271.1089. Anal. Calcd. for C_15_H_12_BF_2_NO: C, 66.46; H, 4.46; N, 5.17%. Found: C, 66.48; H, 4.39, N, 5.20%. Physical and spectral data are in agreement with the literature data [20].
4.5.3. 4-(2,2-Difluoro-6-phenyl-2H-1,3λ4,2λ4-oxazaborinin-3-yl)benzonitrile (5c)
Prepared from enaminone 4c (248 g, 1 mmol), Et_3_N (140 µL, 1 mmol) and BF_3_·Et_2_O (370 μL, 3 mmol), reflux for 4 h, Workup A, CC (EtOAc–hexanes, 1:5 → EtOAc). Yield: 259 mg (88%) of yellow solid, m.p. 207–209 °C, lit. [19] m.p. 208–210 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.08–8.02 (m, 2H), 7.98 (s, 1H), 7.79–7.72 (m, 2H), 7.67–7.58 (m, 3H), 7.53 (t, J = 7.8 Hz, 2H), 6.45 (d, J = 6.0 Hz, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 176.5, 159.4, 146.3, 134.2, 133.7, 132.2, 129.2, 128.4, 124.0, 118.2, 111.8, 94.5. νmax 2230, 1595, 1533, 1489, 1352, 1085, 1025, 1009 cm^−1^. ESI-HRMS: m/z = 296.1045 [M]^+^, C_15_H_11_BF_2_N_2_O requires m/z = 296.1042. Anal. Calcd. for C_15_H_11_BF_2_N_2_O: C, 64.91; H, 3.74; N, 9.46%. Found: C, 64.88; H, 3.75, N, 9.16%. Physical and spectral data are in agreement with the literature data [19].
4.5.4. 2,2-Difluoro-3,4,6-triphenyl-2H-1,3λ4,2λ4-oxazaborinine (5d)
Prepared from enaminone 4i (1.50 g, 5 mmol), Et_3_N (700 µL, 5 mmol) and BF_3_·Et_2_O (1.86 mL, 15 mmol), reflux for 16 h, Workup B. Yield: 1.59 g (92%) of yellow solid, m.p. 195–196 °C, lit. [24] m.p. 195 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.09–8.02 (m, 2H), 7.60–7.53 (m, 1H), 7.49 (m, 2H), 7.38–7.32 (m, 1H), 7.30–7.13 (m, 9H), 6.41 (s, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 172.35, 170.81, 140.66, 135.08, 133.27, 132.95, 130.62, 128.92, 128.87, 128.74, 128.61, 127.86, 127.49, 127.11, 97.00. νmax 1595, 1566, 1506, 1486, 1231, 1107, 1020, 973 cm^−1^. ESI-HRMS: m/z = 347.1400 [M]^+^, C_21_H_16_BF_2_NO requires m/z = 347.1402. Physical and spectral data are in agreement with the literature data [24].
4.6. General Procedure for the Synthesis of Boron Diketonato Complexes 6a–d
These compounds were prepared according to a general procedure in the literature [21]. BF_3_·Et_2_O (1.86 mL, 15 mmol) was added to a solution of 1,3-diketone 1 (10 mmol) in anh. dichloromethane (10 mL), the mixture was stirred at room temperature for 20 h, and volatile components were evaporated in vacuo. The residue was triturated with Et_2_O (10 mL), the precipitate was collected by filtration and washed with Et_2_O (5 mL) to give compound 6.
4.6.1. 2,2-Difluoro-4,6-diphenyl-2H-1,3λ3,2λ4-dioxaborinine (6a)
Prepared from dibenzoylmethane (1c) (2.24 g, 10 mmol) and BF_3_·Et_2_O (1.86 mL, 15 mmol) in anh. dichloromethane (10 mL). Yield: 2.31 g (85%) of light yellow solid, m.p. 194–196 °C, lit. [49] m.p. 195–196 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.19–8.14 (m, 2H), 7.74–7.68 (m, 2H), 7.57 (t, J = 7.9 Hz, 4H), 7.21 (s, 1H). Physical and spectral data are in agreement with the literature data [49].
4.6.2. 2,2-Difluoro-4-methyl-6-phenyl-2H-1,3λ3,2λ4-dioxaborinine (6b)
Prepared from benzoyl acetone (1d) (5.00 g, 30.8 mmol) and BF_3_·Et_2_O (6.00 mL, 46.3 mmol) in anh. dichloromethane (30 mL). Yield: 5.09 g (79%) of white solid, m.p. 125–126 °C, lit. [50] m.p. 120–123 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.08–8.05 (m, 2H), 7.71–7.67 (m, 1H), 7.56–7.51 (m, 2H), 6.58 (s, 1H), 2.42 (s, 3H). νmax 1984, 1898, 1537, 1493, 1356, 1089, 1053, 1018, 777, 706, 679 cm^−1^. ESI-HRMS: m/z = 227.1040 [M+NH_4_]^+^, C_10_H_12_BF_2_NO_2_ requires m/z = 227.020. Physical and spectral data are in agreement with the literature data [50,51].
4.6.3. 2,2-Difluoro-6-(4-methoxyphenyl)-4-methyl-2H-1,3λ3,2λ4-dioxaborinine (6c)
Prepared from (4-methoxylbenzoyl)acetone (1e) (1.92 g, 10 mmol) and BF_3_·Et_2_O (1.86 mL, 15 mmol) in anh. dichloromethane (10 mL). Yield: 1.69 g (70%) of yellowish resin. ^1^H NMR (500 MHz, CDCl_3_) δ 8.06 (d, J = 9.0 Hz, 2H), 7.00 (d, J = 9.0 Hz, 2H), 6.46 (s, 1H), 3.92 (s, 3H), 2.37 (s, 3H). Spectral data are in agreement with the literature data [52].
4.6.4. 2,2-Difluoro-4-methyl-5,6-dihydro-2H-2λ4,3λ3-naphtho[1,2-d][1,3,2]dioxaborinine (6d)
Prepared from 2-acetyl-1-tetralone (1f) (5.00 g, 26.5 mmol) and BF_3_·Et_2_O (5.00 mL, 39.8 mmol) in anh. dichloromethane (25 mL). Yield: 5.14 g (82%) of yellow solid, m.p. 150–153 °C, lit. [28] m.p. 150–154 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.14 (dd, J = 7.8, 0.9 Hz, 1H), 7.56 (td, J = 7.5, 1.3 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.28 (d, J = 7.3 Hz, 1H), 2.98 (t, J = 7.4 Hz, 2H), 2.71 (t, J = 7.4 Hz, 2H), 2.40 (s, 3H). νmax 2961, 2012, 1523, 1431, 1213, 1167, 1138, 1027, 996, 794, 742, 719 cm^−1^. ESI-HRMS: m/z = 253.120 [M+NH_4_]^+^, C_12_H_14_BF_2_NO_2_ requires m/z = 253.050. Physical and spectral data are in agreement with the literature data [28].
4.7. General Procedure for the Synthesis of 1,5-Diketones 7a–f
A 5 mL vial with a screw cap and septum was charged with boron β-diketonate 6 (0.5 mmol), alkene (0.5–5 mmol), acetophenone (1.5 mg, 12 μmol), and dichloromethane or THF (5 mL). The vial was stopped and carefully degassed and purged with nitrogen using the freeze–pump–thaw technique. Then, the vial was mounted into a photoreactor and irradiated with UV-A light (365 nm) at 20 °C for 20 h. The reaction mixture was diluted with dichloromethane (20 mL) and washed with 1 M aq. HCl (15 mL), saturated aq. NaHCO_3_ (15 mL), and brine (15 mL). The organic phase was dried over anh. sodium sulfate, filtered, and the filtrate was evaporated in vacuo. The residue was purified by CC (ethyl acetate–petroleum ether). Fractions containing the product were combined and evaporated in vacuo to give the product 7.
4.7.1. 2-(2-Benzoylcyclohexyl)-1-phenylethan-1-one (7a)
Prepared from boron β-diketonate 6a (137 mg, 0.5 mmol), cyclohexene (164 mg, 2 mmol), and acetophenone (1.5 mg, 12 μmol) in dichloromethane (5 mL), CC (EtOAc–hexanes, 1:50). Yield: 28 mg (18%) of white resin. ^1^H NMR (500 MHz, CDCl_3_) δ 8.01–7.96 (m, 4H), 7.60–7.42 (m, 6H), 3.32 (td, J = 11.2, 3.4 Hz, 1H), 3.13 (dd, J = 13.8, 2.7 Hz, 1H), 2.48 (dd, J = 13.8, 10.0 Hz, 1H), 2.54 (qt*, J* = 10.2, f3.2 Hz, 1H), 2.01–1.95 (m, 1H), 1.91–1.85 (m, 1H), 1.84–1.79 (m, 1H), 1.78–1.71 (m, 1H), 1.44–1.15 (m, 4H). ^13^C NMR (126 MHz, CDCl_3_) δ 203.7, 199.9, 137.3, 137.0, 133.0, 132.9, 128.8, 128.6, 128.3, 128.2, 46.6, 39.0, 33.7, 29.4, 25.9, 23.8, 23.0. νmax 3058, 2927, 2855, 1674, 1596, 1579, 1447, 1279, 1251, 1212, 117, 1002, 981, 944, 751, 690, 663 cm^−1^. ESI-HRMS: m/z = 307.1695 [MH]^+^, C_21_H_23_O_2_ requires m/z = 307.1693. Physical and spectral data are in agreement with the literature data [53].
4.7.2. 2-(2-Benzoylcyclopentyl)-1-phenylethan-1-one (7b)
Prepared from boron β-diketonate 6a (68 mg, 0.25 mmol), cyclopentene (130 μL, 1.5 mmol), and acetophenone (0.75 mg, 6 μmol) in dichloromethane (2.5 mL), CC (EtOAc–hexanes, 1:50). Yield: 16 mg (22%) of white resin. ^1^H NMR (500 MHz, CDCl_3_) δ 8.00–7.93 (m, 4H), 7.58–7.51 (m, 2H), 7.47 (t, J = 7.7 Hz, 2H), 7.44 (t, J = 7.7 Hz, 2H), 3.52 (dt, J = 9.2, 7.8 Hz,1H), 3.19 (dd, J = 15.0, 5.5 Hz, 1H), 3.00 (pd, J = 8.3, 5.5 Hz, 1H), 2.87 (dd, J = 15.1, 8.5 Hz, 1H), 2.20–2.12 (m, 1H), 2.11–2.02 (m, 1H), 1.83–1.67 (m, 3H), 1.43 (dq, J = 12.5, 8.1 Hz, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 202.4, 199.9, 137.3, 137.0, 133.1, 133.0, 128.7, 128.7, 128.6, 128.4, 52.7, 43.9, 38.9, 32.5, 31.5, 24.8. νmax 3061, 2949, 2869, 1677, 1596, 1579, 1448, 1276, 1220, 1001, 752, 690 cm^−1^. ESI-HRMS: m/z = 293.1536 [MH]^+^, C_20_H_21_O_2_ requires m/z = 293.1536. Spectral data are in agreement with the literature data [54].
4.7.3. 1-(2-Benzoylcyclohexyl)propan-2-one (7c)
Prepared from boron β-diketonate 6b (106 mg, 0.5 mmol), cyclohexene (250 mg, 3 mmol), and acetophenone (1.5 mg, 12 μmol) in dichloromethane (5 mL), CC (EtOAc–hexanes, 1:50). Yield: 16 mg (13%) of white resin. ^1^H NMR (500 MHz, CDCl_3_) δ 7.91–7.86 (m, 2H), 7.53 (tt, J = 7.3, 1.3 Hz, 1H), 7.44 (t, J = 7.6 Hz, 2H), 3.63 (dt, J = 8.2, 4.0 Hz, 1H), 2.60–2.52 (m, 1H), 2.52 (dd, J = 17.1, 7.2 Hz, 1H), 2.45 (dd, J = 17.1, 5.5 Hz, 1H), 2.00 (s, 3H), 1.87–1.78 (m, 2H), 1.71–1.34 (m, 6H). ^13^C NMR (126 MHz, CDCl_3_) δ 208.2, 203.7, 137.0, 133.0, 128.8, 128.3, 46.3, 44.0, 32.8, 30.7, 29.7, 25.7, 23.8, 22.8. νmax 2927, 2857, 1712, 1673, 1447, 1356, 1253, 1214, 1161, 1002, 976, 759, 699 cm^−1^. ESI-HRMS: m/z = 245.1534 [MH]^+^, C_16_H_21_O_2_ requires m/z = 245.1536. Physical and spectral data are in agreement with the literature data [53].
4.7.4. 1-[2-(4-Methoxybenzoyl)cyclohexyl]propan-2-one (7d)
Prepared from boron β-diketonate 6c (48 mg, 0.20 mmol), cyclohexene (120 μL, 1.2 mmol), and acetophenone (0.75 mg, 6 μmol) in THF (2.5 mL), CC (EtOAc–hexanes, 1:10). Yield: 5 mg (9%) of brown resin. ^1^H NMR (500 MHz, CDCl_3_) δ 7.95 (br d, J = 7.7 Hz, 2H), 6.95 (br d, J = 7.5 Hz, 2H), 3.87 (s, 3H), 3.18 (br t, J = 9.8 Hz, 1H), 2.43–2.31 (m, 2H), 2.20–2.00 (m, 1H), 2.07 (s, 3H), 1.95–1.84 (m, 2H), 1.83–1.72 (m, 2H), 1.43–1.31 (m, 3H), 1.19–1.07 (m, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 208.7, 202.3, 163.7, 130.7, 130.0, 114.0, 55.6, 49.8, 49.2, 35.3, 31.7, 31.5, 30.1, 26.1, 25.8. ν_max_ 2930, 2856, 1708, 1671, 1598, 1510, 1358, 1254, 1169, 1114, 1028, 839, 758 cm^−1^. ESI-HRMS: m/z = 275.1643 [MH]^+^, C_17_H_23_O_3_ requires m/z = 275.1642.
4.7.5. 4-Acetyl-1,2,3,3a,4,5,6,11a-octahydro-11H-benzo[a]cyclopenta[d][8]annulen-11-one (7e)
Prepared from boron β-diketonate 6d (59 mg, 0.25 mmol), cyclopentene (130 μL, 1.5 mmol), and acetophenone (1.5 mg, 12 μmol) in dichloromethane (2.5 mL), CC (EtOAc–hexanes, 1:10). Yield: 18 mg (28%) of yellowish resin. ^1^H NMR (500 MHz, CDCl_3_) δ 8.02 (dd, J = 7.9, 1.4 Hz, 1H), 7.46 (td, J = 7.4, 1.5 Hz, 1H), 7.35 (td, J = 7.9, 1.1 Hz, 1H), 7.22 (dd, J = 7.5, 0.6 Hz, 1H), 3.97 (td, J = 7.8, 2.4 Hz, 1H), 3.77 (td, J = 14.2, 5.2 Hz, 1H), 2.94 (ddd, J = 14.9, 5.2, 2.9 Hz, 1H), 2.48–2.29 (m, 3H), 2.04 (s, 3H), 2.02–1.86 (m, 2H), 1.82–1.70 (m, 3H), 1.61–1.50 (m, 1H), 1.31–1.18 (m, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 212.1, 204.5, 140.1, 139.4, 133.2, 132.3, 130.1, 127.2, 52.1, 51.3, 46.3, 34.4, 31.7, 31.0, 30.6, 27.8, 23.5. νmax 2941, 2853, 1693, 1663, 1595, 1464, 1443, 1358, 1345, 1306, 1277, 1202, 1179, 759 cm^−1^. ESI-HRMS: m/z = 257.1540 [MH]^+^, C_17_H_21_O_2_ requires m/z = 257.1536.
4.7.6. 5-Acetyl-1,3,4,4a,5,6,7,12a-octahydrodibenzo[a,d][8]annulen-12(2H)-one (7f)
Prepared from boron β-diketonate 6d (236 mg, 1.0 mmol), cyclohexene (164 mg, 2 mmol), and acetophenone (1.5 mg, 12 μmol) in dichloromethane (5 mL), CC (EtOAc–hexanes, 1:50). Yield: 80 mg (30%) of yellowish resin. ^1^H NMR (500 MHz, CDCl_3_) δ 7.54 (br d, J = 7.4 Hz, 1H), 7.39 (td, J = 7.5, 1.4 Hz, 1H), 7.30 (td, J = 7.6, 0.9 Hz, 1H), 7.13 (br d, J = 7.6 Hz, 1H), 3.21 (dt, J = 15.7, 9.7 Hz, 1H), 3.15 (br s, 1H), 2.94 (dt, J = 15.7, 5.3 Hz, 1H), 2.57–2.42 (m, 2H), 2.11–2.00 (m, 1H), 2.06 (s, 3H), 1.97–1.88 (m, 2H), 1.85–1.71 (m, 2H), 1.60–1.50 (m, 1H), 1.48–1.32 (m, 2H), 1.29–1.21 (m 1H), 1.13–1.02 (m, 1H). ^13^C NMR (126 MHz, CDCl_3_) δ 211.6, 207.9, 140.8, 137.9, 131.3, 130.2, 128.4, 126.9, 52.1, 51.8, 38.4, 32.2, 30.5, 30.0, 27.2, 26.2, 24.4, 23.4. νmax 2914, 1848, 1703, 1659, 1230, 954, 925, 764, 682 cm^−1^. ESI-HRMS: m/z = 271.1690 [MH]^+^, C_18_H_23_O_2_ requires m/z = 271.1693.
4.8. General Procedure for the Synthesis of Compounds 8a–d
A mixture of enaminone 4 (1 mmol) and anh. acetonitrile (5 mL) was stirred at 0 °C (ice bath) for 5 min. Then, Boc_2_O (327 mg, 1.5 mmol) and DMAP (12 mg, 0.1 mmol) were added, and the mixture was stirred at 0 °C for 10 min. Ice bath was removed, and the mixture was stirred at room temperature for 16 h. Volatile components were evaporated in vacuo and the residue was purified by CC (EtOAc–hexanes). Fractions containing the product were combined and evaporated in vacuo to give compound 8.
4.8.1. tert-Butyl (E)-(3-oxo-3-phenylprop-1-en-1-yl)(phenyl)carbamate (8a)
Prepared from enaminone 4b (224 mg, 1 mmol), CC (EtOAc–hexanes, 1:10). Yield: 297 mg (92%) of yellow solid, m.p. 109–110 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.60 (d, J = 13.7 Hz, 1H), 7.74–7.69 (m, 2H), 7.53–7.41 (m, 4H), 7.40–7.34 (m, 2H), 7.23–7.19 (m, 2H), 5.79 (d, J = 13.7 Hz, 1H), 1.48 (s, 9H). ^13^C NMR (126 MHz, CDCl_3_) δ 190.5, 152.0, 145.4, 138.8, 137.9, 132.2, 129.9, 128.8, 128.5, 128.2, 128.1, 105.2, 83.8, 28.1. νmax 2983, 2969, 1720 (C=O), 1655 (C=O), 1564, 1345, 1270, 1141 cm^−1^. ESI-HRMS: m/z = 324.1589 [M+H]^+^, C_20_H_22_NO_3_ requires m/z = 324.1594.
4.8.2. tert-Butyl (E)-allyl(3-(4-chlorophenyl)-3-oxoprop-1-en-1-yl)carbamate (8b)
Prepared from enaminone 4e (444 mg, 2 mmol), CC (EtOAc–hexanes, 1:5). Yield: 644 mg (100%) of yellow solid, m.p. 83–84 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.40 (d, J = 13.7 Hz, 1H), 7.82 (d, J = 8.6 Hz, 2H), 7.42 (d, J = 8.5 Hz, 2H), 6.20 (d, J = 13.8 Hz, 1H), 5.81 (ddt, J = 17.2, 10.3, 5.1 Hz, 1H), 5.25 (dq, J = 10.5, 1.4 Hz, 1H), 5.19 (dq, J = 17.1, 1.7 Hz, 1H), 4.29 (dt, J = 5.3, 1.8 Hz, 2H), 1.54 (s, 9H). ^13^C NMR (126 MHz, CDCl_3_) δ 189.0, 152.1, 144.2, 138.58, 137.4, 131.3, 129.6, 128.9, 117.2, 102.7, 83.9, 47.2, 28.2. νmax 3118, 3010, 2928, 1715 (C=O), 1653 (C=O), 1590, 1577, 1560, 1489, 1456, 1409, 1393, 1368, 1359, 1320, 1283, 1244, 1204, 1172, 1135, 1090, 1045, 1011, 981, 956, 944, 933, 912, 886, 845, 832, 816, 771, 742, 680 635 cm^−1^. ESI-HRMS: m/z = 322.1203 [M+H]^+^, C_17_H_21_ClNO_3_ requires m/z = 322.1204.
4.8.3. tert-Butyl (E)-(3-oxo-3-phenylprop-1-en-1-yl)(pyridin-2-yl)carbamate (8c)
Prepared from enaminone 4f (224 mg, 1 mmol), CC (EtOAc–hexanes, 1:5). Yield: 324 mg (100%) of yellow solid, m.p. 105–107 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 8.68 (ddd, J = 4.8, 2.0, 0.8 Hz, 1H), 8.50 (d, J = 14.0 Hz, 1H), 7.90 (td, J = 7.7, 2.0 Hz, 1H), 7.76–7.71 (m, 2H), 7.51–7.45 (m, 1H), 7.43–7.36 (m, 3H), 7.28 (dt, J = 7.8, 1.0 Hz, 1H), 5.81 (d, J = 13.9 Hz, 1H), 1.49 (s, 9H). ^13^C NMR (126 MHz, CDCl_3_) δ 190.5, 151.5, 151.2, 150.1, 144.1, 139.1, 138.7, 132.2, 128.5, 128.1, 124.0, 123.4, 105.5, 84.2, 28.1. νmax 3012, 2979, 1720 (C=O), 1655 (C=O), 1563, 1467, 1213, 1098 cm^−1^. ESI-HRMS: m/z = 325.1540 [M+H]^+^, C_19_H_21_N_2_O_3_ requires m/z = 325.1547.
4.8.4. tert-Butyl (E/Z)-(3-oxo-1,3-diphenylprop-1-en-1-yl)(phenyl)carbamate (8d)
Prepared from enaminone 4i (299 mg, 1 mmol), CC (EtOAc–hexanes, 1:5). Yield: 352 mg (88%) of light yellow solid, m.p. 113–124 °C, E/Z = 4:1. ^1^H NMR (500 MHz, CDCl_3_) δ major isomer 7.88 (d, J = 7.5 Hz, 2H), 7.69–7.60 (m, 2H), 7.60–7.50 (m, 1H), 7.48–7.35 (m, 5H), 7.28–7.21 (m, 1H), 7.19–7.10 (m, 3H), 7.06–6.98 (m, 1H), 6.91 (s, 1H), 1.26 (s, 9H); minor isomer 7.76 (dd, J = 8.3, 1.2 Hz, 2H), 7.47–7.37 (m, 7H), 7.30 (t, J = 7.7 Hz, 2H), 7.27–7.21 (m, 1H), 7.20–7.10 (m, 3H), 6.37 (s, 1H), 1.20 (s, 9H). ^13^C NMR (126 MHz, CDCl_3_) δ major isomer ^13^C NMR (126 MHz, CDCl_3_) δ 189.8, 153.1, 151.4, 141.7, 138.4, 132.9, 130.1, 129.4, 128.9, 128.6, 128.5, 128.4, 127.4, 127.0, 125.3, 119.0, 81.9, 28.0; minor isomer 192.8, 154.4, 153.5, 143.0, 138.2, 137.1, 132.7, 130.1, 129.5, 128.9, 128.8, 128.3, 128.2, 126.7, 125.5, 121.4, 82.4, 27.8. νmax 2977, 1713 (C=O), 1660 (C=O), 1595, 1491, 1448, 1367, 1339, 1269, 1246, 1153, 1014, 847, 771, 741, 692 cm^−1^. Anal. Calcd. for C_26_H_25_NO_3_·⅙H_2_O: C, 77.52; H, 6.69, N, 3.45%. Found: C, 77.59; H, 6.34, N, 3.48%.
4.9. Synthesis of (Z)-{2-Benzoyl-3-[(4-cyanophenyl)amino]acryloyl}sulfamoyl chloride (9)
Under argon, chlorosulfonyl isocyanate (CSI) (130 μL, 1.5 mmol) was added to a suspension of enaminone 4c (248 mg, 1 mmol) in anhydrous Et_2_O (10 mL) at 0 °C (ice-bath) and the mixture was stirred at 0 °C for 15 minutes. Then, the ice bath was removed and stirring was continued at room temperature (~20 °C) for 1 h. The precipitate was collected by filtration, washed with anh. Et_2_O (3 × 5 mL) and dried in vacuo over NaOH pellets for 12 h. Yield: 310 mg (80%) of yellowish solid, m.p. 150 °C (decomp.). ^1^H NMR (600 MHz, CDCl_3_) δ 13.84 (s, 1H), 12.14 (d, J = 13.4 Hz, 1H), 8.26 (d, J = 13.4 Hz, 1H), 7.69 (br d, J = 8.7 Hz, 2H), 7.67–7.62 (m,1H), 7.59–7.54 (m, 4H), 7.13 (br d, J = 8.7 Hz, 2H). ^13^C NMR (150 MHz, CDCl_3_) δ 196.1, 165.7, 156.4, 141.2, 137.6, 134.3, 132.5, 129.0, 128.7, 118.2, 117.8, 110.2, 102.6. νmax 2223 (C≡N), 1775 (C=O), 1717 (C=O), 1672 (C=O), 1629 (C=O), 1588, 1567, 1440, 1414, 1318, 1275, 1177, 907, 841, 814, 762, 734, 709, 661 cm^−1^.
4.10. Allylation of Enaminone 4b. Synthesis of Compounds 10–12
Under argon, allyl bromide (175 μL, 2 mmol) was added to a stirred mixture of enaminone 4b (224 mg, 1 mmol), K_2_CO_3_ (500 mg, 3.6 mmol), NaI (12 mg, 0.08 mmol), and anh. acetonitrile (25 mL). The reaction mixture was then stirred under reflux under argon for 16 h. Volatile components were evaporated in vacuo to give a crude mixture of compounds 10, 11, and 12. The residue was purified by CC (EtOAc–hexanes, 1:5). Compound 10 eluted first, followed by elution of compound 12 and compound 11. Fractions containing the products 10, 11, and 12 were combined and evaporated in vacuo to give compounds 10, 11, and 12.
4.10.1. (E)-2-Allyl-1-phenyl-2-[(phenylimino)methyl]pent-4-en-1-one (10)
Yield: 135 mg (44%) of yellow–orange oil. ^1^H NMR (500 MHz, CDCl_3_) δ 8.00 (s, 1H), 7.93–7.89 (m, 2H), 7.53–7.49 (m, 1H), 7.44–7.39 (m, 2H), 7.37–7.32 (m, 2H), 7.24–7.19 (m, 1H), 7.04–6.99 (m, 2H), 5.74 (ddt, J = 17.5, 10.2, 7.4 Hz, 2H), 5.12–5.02 (m, 4H), 2.94 (dt, J = 7.4, 1.2 Hz, 4H). ^13^C NMR (126 MHz, CDCl_3_) δ 200.0, 166.7, 151.7, 137.2, 132.6, 132.4, 129.2, 129.2, 128.4, 126.0, 120.4, 119.2, 59.9, 38.3. ESI-HRMS: m/z = 304.1687 [M+H]^+^, C_21_H_22_NO requires m/z = 304.1696.
4.10.2. (E)-3-[Allyl(phenyl)amino]-1-phenylprop-2-en-1-one (11)
Yield: 106 mg (40%) of yellow–orange oil. ^1^H NMR (500 MHz, CDCl_3_) δ 8.24 (d, J = 12.8 Hz, 1H), 7.90 (dt, J = 7.1, 1.4 Hz, 2H), 7.51–7.46 (m, 1H), 7.46–7.41 (m, 2H), 7.40–7.35 (m, 2H), 7.26–7.23 (m, 2H), 7.19 (td, J = 7.3, 1.1 Hz, 1H), 6.10 (d, J = 12.9 Hz, 1H), 5.94 (ddt, J = 17.2, 10.5, 4.8 Hz, 1H), 5.34–5.26 (m, 2H), 4.39 (dt, J = 4.2, 1.9 Hz, 2H). ^13^C NMR (126 MHz, CDCl_3_) δ 189.6, 149.2, 140.1, 131.5, 131.1, 129.7, 129.3, 128.4, 127.8, 125.3, 117.8, 115.6, 97.7, 53.5. ESI-HRMS: m/z = 264.1375 [M+H]^+^, C_18_H_18_NO requires m/z = 264.1383.
4.10.3. (E)-2-{[Allyl(phenyl)amino]methylene}-1-phenylpent-4-en-1-one (12)
Yield: 21 mg (7%) of yellow–orange oil. ^1^H NMR (500 MHz, CDCl_3_) δ 7.57–7.54 (m, 2H), 7.42–7.38 (m, 3H), 7.35 (s, 1H), 7.31–7.26 (m, 2H), 7.14–7.09 (m, 1H), 7.06–7.02 (m, 2H), 5.96–5.79 (m, 2H), 5.21 (d, J = 2275.4 Hz, 2H), 4.95 (dd, J = 99.1, 53.9 Hz, 2H), 4.31 (dt, J = 4.6, 1.9 Hz, 2H), 3.12 (dt, J = 5.4, 1.9 Hz, 2H). ^13^C NMR (126 MHz, CDCl_3_) δ 197.4, 151.5, 146.0, 141.2, 137.1, 133.5, 129.9, 129.2, 128.6, 128.0, 125.0, 121.9, 117.2, 114.3, 114.3, 55.8, 28.9. ESI-HRMS: m/z = 304.1691 [M+H]^+^, C_21_H_22_NO requires m/z = 304.1696.
4.11. Synthesis of 1-allyl-3,5-bis(4-chlorobenzoyl)-1,4-dihydropyridine (13)
A 5 mL flask was charged with compound 4e (222 mg, 1 mmol) and the flask was cooled to 0 °C (ice bath). Formic acid (700 μL), 37% aq. formaldehyde (1.35 mL), and water (1 mL) were added at 0 °C (ice bath), the flask was equipped with a reflux condenser, and the mixture was stirred at 90 °C for 48 h. The reaction mixture was cooled to room temperature, diluted with 6 M hydrochloric acid (20 mL), and the product was extracted with dichloromethane (3 × 15 mL). The combined organic phase was evaporated in vacuo and the residue was purified by CC (CH_2_Cl_2_–MeOH, 50:1). Fractions containing the product were combined and evaporated in vacuo to give 13. Yield: 42 mg (22%) of brownish solid, m.p. 156–159 °C. ^1^H NMR (500 MHz, CDCl_3_) δ 7.49 (d, J = 8.4 Hz, 4H), 7.40 (d, J = 8.4 Hz, 4H), 6.65 (s, 2H), 5.79 (ddt, J = 17.0, 10.5, 5.3 Hz, 1H), 5.30 (dd, J = 8.6, 1.8 Hz, 1H), 5.25 (dd, J = 17.2, 1.0 Hz, 1H), 3.82 (dt, J = 5.5, 1.6 Hz, 2H), 3.51 (s, 2H). ^13^C NMR (150 MHz, CDCl_3_) δ 193.0, 143.1, 137.4, 137.1, 132.1, 129.9, 128.6, 119.2, 115.7, 56.8, 21.4. νmax 3088, 2827, 2050, 1664, 1610, 1584, 1572, 1483, 1451, 1396, 1374, 1289, 1258, 1201, 1170, 1136, 1087, 1012, 976, 961, 932, 911, 845, 826, 779, 743, 683, 654, 628 cm^−1^. ESI-HRMS: m/z = 398.0691 [M+H]^+^, C_22_H_28_Cl_2_NO_2_ requires m/z = 398.0709.
5. Conclusions
An array of acetophenone-derived enaminones were prepared via the transamination of N,N-dimethylenaminones. The corresponding BF_2_-β-ketoiminates as well as BF_2_-β-diketonates were then prepared and tested in De Mayo reactions with cyclic alkenes. While BF_2_-β-diketonates gave the desired products in low yields, BF_2_-β-ketoiminates remained unconverted. The photochemical transformation of tricyclic BF_2_-acetyltetralone-complex resulted in ring expansion to give novel diannulated cyclooctane derivatives, which would be difficult to obtain using conventional cyclization methods. Additionally, N-Boc, α-chlorosulfonylated, C,C-diallylated, N-monoallyated, and C,N-diallylated derivatives of enaminones were prepared. While their attempted photochemical transformations failed, interesting mechanistic insights were made into allylation of enaminones as well as their conversion to 1,4-dihydropyridine derivative.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Greenhill J.V. Enaminones Chem. Soc. Rev.1977627729410.1039/cs 9770600277 · doi ↗
- 2Stanovnik B. Svete J. Synthesis of Heterocycles from Alkyl 3-(Dimethylamino)propenoates and Related Enaminones Chem. Rev.20041042433248010.1021/cr 020093 y 15137796 · doi ↗ · pubmed ↗
- 3Šimůnek P. Maháček V. The structure and tautomerism of azo coupled β-Enaminones Dyes Pigment.20108618720510.1016/j.dyepig.2010.01.004 · doi ↗
- 4Chattopadhyay A.K. Hanessian S. Cyclic enaminones. Part I: Stereocontrolled synthesis using diastereoselective and catalytic asymmetric methods Chem. Commun.201551164371644910.1039/C 5CC 05891 K 26490402 · doi ↗ · pubmed ↗
- 5Chattopadhyay A.K. Hanessian S. Cyclic enaminones. Part II: Applications as versatile intermediates in alkaloid synthesis Chem. Commun.201551164501646710.1039/C 5CC 05892 A 26490499 · doi ↗ · pubmed ↗
- 6Huang J. Yu F. Recent Advances in Organic Synthesis Based on N,N-Dimethyl Enaminones Synthesis 20215358761010.1055/s-0040-1707328 · doi ↗
- 7Amaye I.J. Haywood R.D. Mandzo E.M. Wirick J.J. Jackson-Ayotunde P.L. Enaminones as building blocks in drug development: Recent advances in their chemistry, synthesis, and biological properties Tetrahedron 20218313198410.1016/j.tet.2021.131984 · doi ↗
- 8Wang Z. Zhao B. Liu Y. Wan J.-P. Recent Advances in Reactions Using Enaminone in Water or Aqueous Medium Adv. Synth. Catal.20223641508152110.1002/adsc.202200144 · doi ↗