Oral Cancer-Derived miR-762 Suppresses T-Cell Infiltration and Activation by Horizontal Inhibition of CXCR3 Expression
Hsuan-Yu Peng, Chia-Wei Chang, Ping-Hsiu Wu, Li-Jie Li, Yu-Lung Lin, Michael Hsiao, Jang-Yang Chang, Peter Mu-Hsin Chang, Hsin-Lun Lee, Wei-Min Chang

TL;DR
This study shows that miR-762 from oral cancer cells suppresses T-cell activity by reducing CXCR3, contributing to an immunosuppressive tumor environment.
Contribution
The study identifies miR-762 as a novel RNA molecule that horizontally inhibits T-cell function through CXCR3 suppression in oral cancer.
Findings
miR-762 suppresses T-cell migration and cytotoxicity by inhibiting CXCR3 expression.
Horizontal transfer of miR-762 from cancer cells to T cells disrupts AKT activation and T-cell activation markers.
miR-762 reduces IL-12 secretion and T-cell proliferation in co-culture systems.
Abstract
Oral squamous cell carcinoma (OSCC) is an immune-cold tumor characterized by an immunosuppressive microenvironment with low cytotoxic activity to eliminate tumor cells. Tumor escape is one of the initial steps in cancer development. Understanding the underlying mechanisms of cancer escape can help researchers develop new treatment strategies. In this study, we prove the oral oncogenic miR-762 can suppress T-cell recruitment and cytotoxic activation in the tumor microenvironment (TME) through horizontal transmission from OSCC cells to adaptive immune T cells. Public database analysis and quantitative real-time polymerase chain reaction (qRT-PCR) were used to determine the prognosis and expression of miR-762 in OSCC. T-cell activation by flow cytometry, qRT-PCR, IL-12 secretion, and T-cell recruitment and cytotoxicity abilities were conducted in the miR-762 manipulation T-cell and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
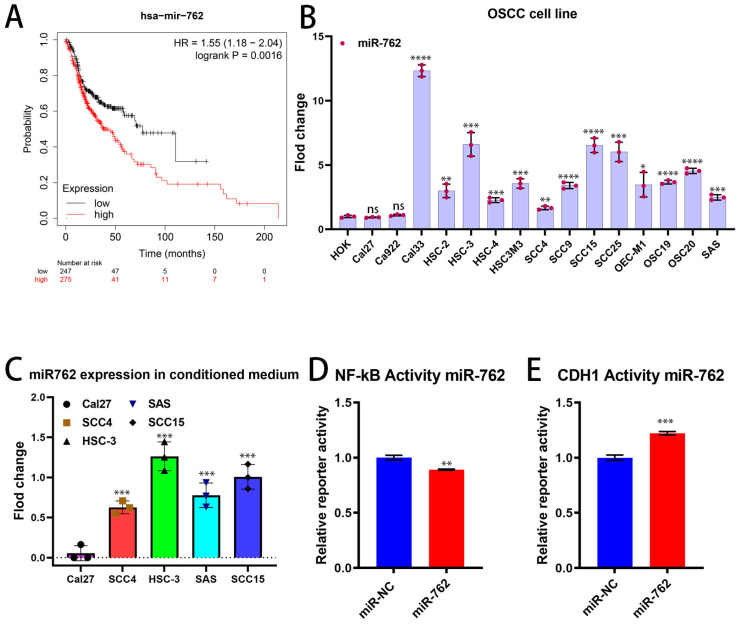 Figure 1
Figure 1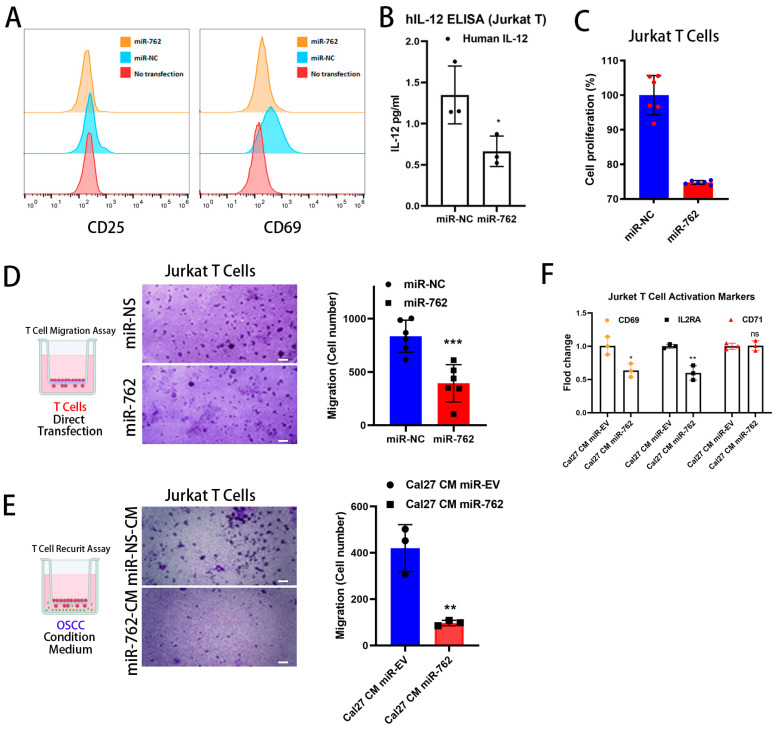 Figure 2
Figure 2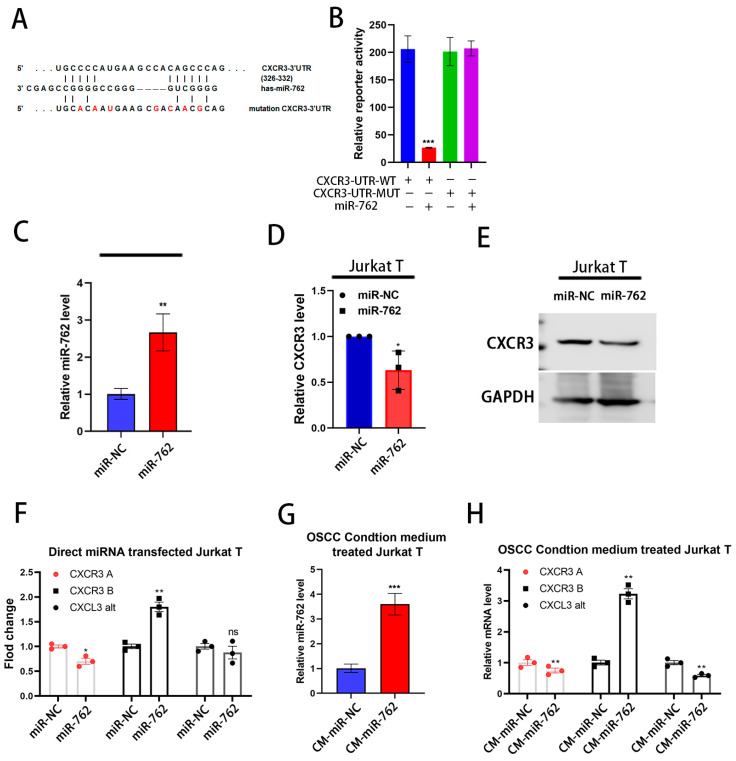 Figure 3
Figure 3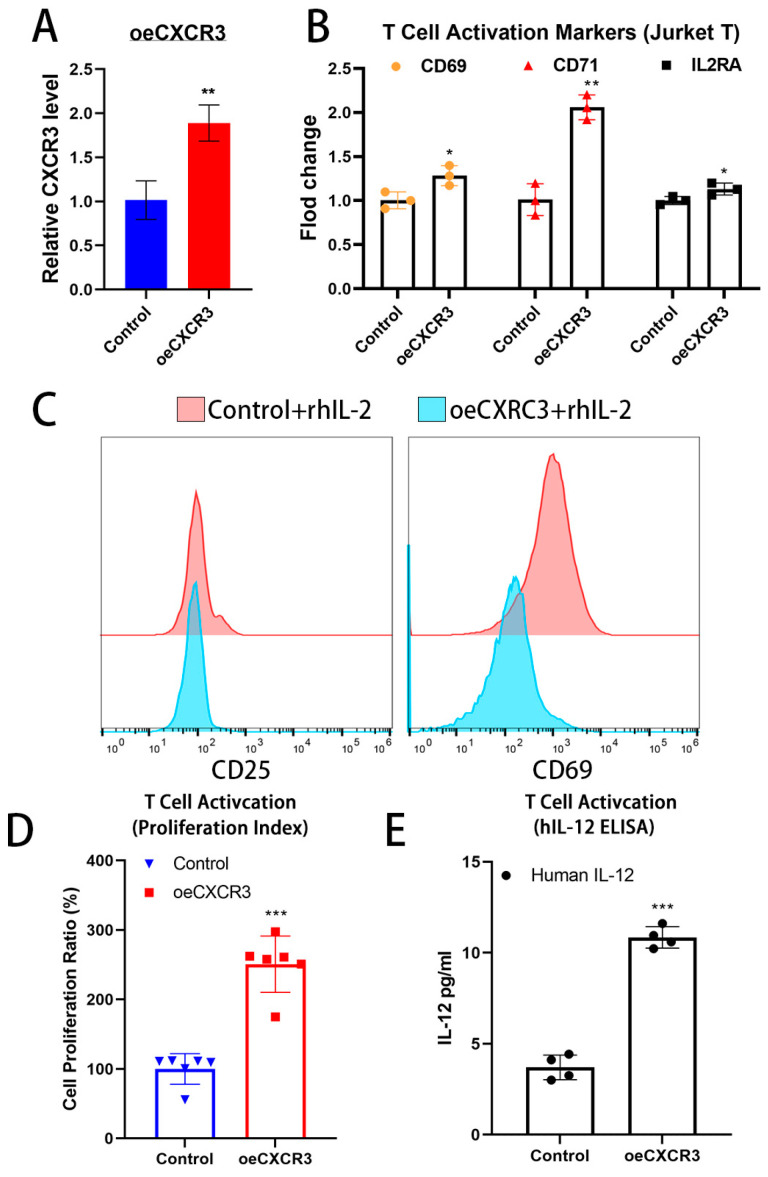 Figure 4
Figure 4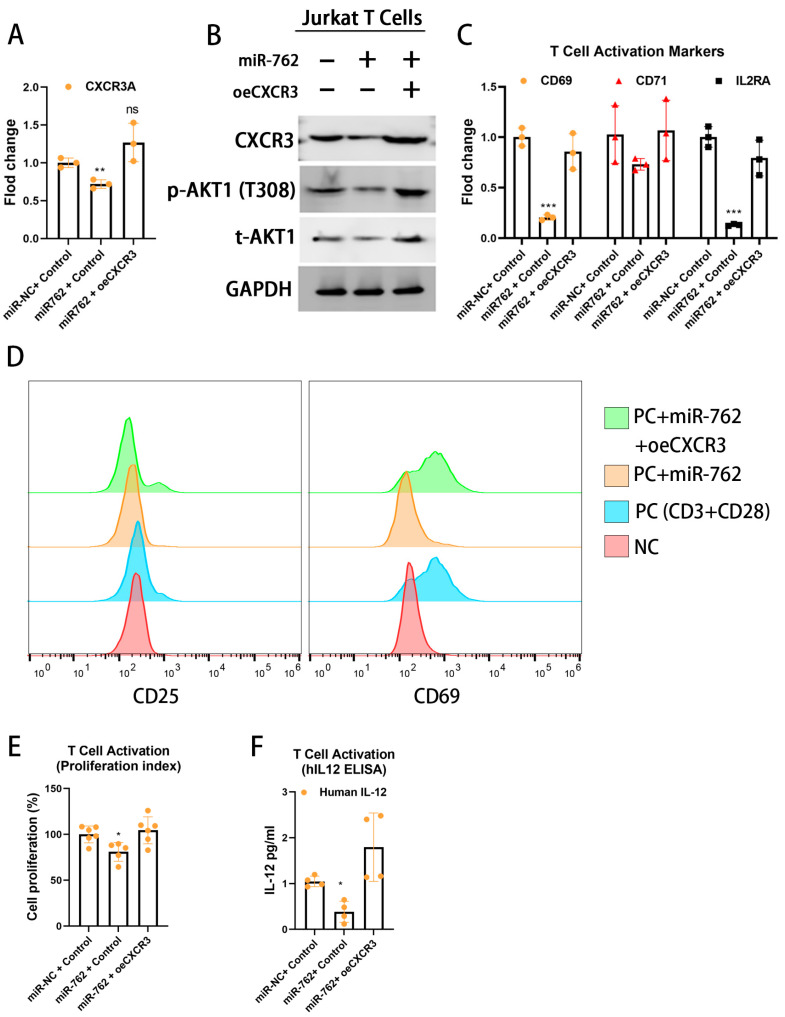 Figure 5
Figure 5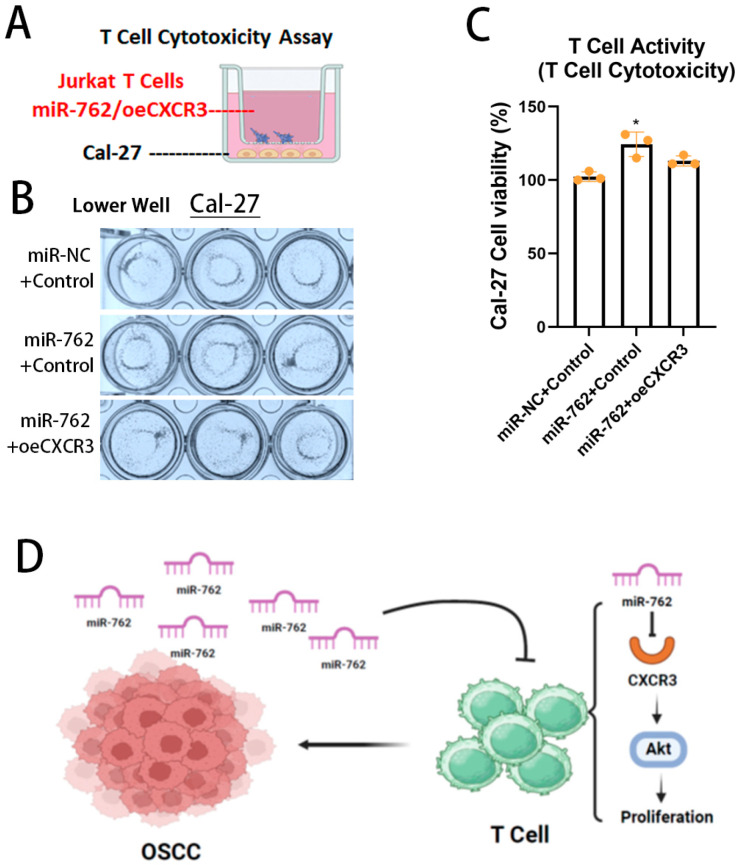 Figure 6
Figure 6- —Taipei Medical University Research Center of Cancer Translational Medicine
- —Higher Education Sprout Project, Taiwan Ministry of Education
- —Taipei Medical University Hospital, Taipei Medical University
- —Taipei Veterans General Hospital
- —Taipei Medical University
- —National Science and Technology Council, Taiwan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmunotherapy and Immune Responses · Chemokine receptors and signaling · MicroRNA in disease regulation
1. Introduction
Cancer is the deadliest disease, with an estimated 18.1 million new cases and 10 million deaths annually. In the USA, 1.9 million new cases and 609,820 deaths are expected each year [1]. Oral cancer is severely impacting patients’ quality of life and causing socioeconomic impacts [2]. The host immune system plays a crucial role in eliminating cancer cells during tumor initiation [3]. Effector immune cells, like T cells, γδT cells, and natural killer (NK) cells, are responsible for elimination [4]. These cells must infiltrate into the tumor microenvironment (TME). However, OSCC employs several strategies to evade the immune system, including expression of inflammatory cytokines, suppression of cytotoxic CD8 lymphocytes, downregulation of antigen processing machinery, generation of specific inhibitory lymphocytes, and expression of immune checkpoint ligands and receptors. These mechanisms increase OSCC resistance to cytotoxic T cells and inhibit T-cell function, facilitating tumor initiation and growth [5]. Understanding immune-escape mechanisms is essential for developing new treatment strategies for OSCC.
Chemokines are small cytokines that direct immune cell recruitment. The chemokine family consists of about 50 ligands, 20 G protein-coupled receptors (GPCRs), and 4 atypical chemokine receptors (ACKRs) [6]. They play a key role in cancer immunity by attracting immune cells to the tumor microenvironment (TME). CXCR3, a critical receptor, aids in T and NK cell recruitment and macrophage polarization [7]. Its expression is linked to the progression of cancers like breast, colorectal, and pancreatic. High levels of CXCR3 ligands in pancreatic and colorectal cancers are associated with poor survival, making it a potential prognostic marker [8]. The diverse functions of CXCR3 underscore its potential as a target for immunotherapy and its significance in cancer and immune regulation.
The TME is a dynamic ecosystem involving cancer cells and host cells like immune cells, fibroblasts, and endothelial cells [9]. In tumor initiation, dysregulated immune cell distribution shapes the TME immune landscape [10]. TME immune status affects patient prognosis and the outcomes of treatment. The TME EVs harbor proteins and free nucleic acids, such as enzymes, receptors, mRNA, non-coding RNA, and microRNA, and serve as colonial rulers for horizontal transmission between cancer donors and host recipient T cells [11]. MicroRNAs, known for their stability and gene regulation capabilities, play a critical role in TME [12]. Aberrant microRNA expression has been reported in OSCC tumorigenesis and progression [13]. miR-762, an oncogenic microRNA, contributes to OSCC and is a poor prognostic marker in colon cancer due to WNT signaling [14]. Previously, we found that miR-762 is a serum prognostic marker and contributes to colon cancer progression through WNT signaling [15]. Here, we found that miR-762 suppresses the immune response in OSCC by targeting CXCR3 and hindering cytotoxic T-cell recruitment and activation.
2. Results
2.1. miR-762 Significantly Associated with the Poor Prognosis of OSCC
To investigate the relationship between miR-762 and clinical manifestations in OSCC patients by using the Kaplan–Meier plotter database. The high-miR-762 group had significantly shorter overall survival (OS, Figure 1A). The median OS was 77.3 months in the low-miR-762 group versus 37.77 months in the high-miR-762 group. We also found that miR-762 levels are high in OSCC cell lines compared to normal keratinocyte cells (Figure 1B). OSCC is an immune cold tumor, and OSCC typically has an immunosuppressive microenvironment, involving immune checkpoint proteins, environmental immunosuppressive factors, and an imbalance between immunosuppressive and effective immune cell compositions [3]. This leads to diminished T-cell numbers and activity in the tumor, resulting in poor prognosis for OSCC patients [16]. We found that miR-762 is secreted by OSCC cells (Figure 1C), with Cal-27 having the lowest secretion of miR-762 among the five OSCC cell lines (Cal-27, SCC-4, HSC-3, SAS, and SCC-15). Secreted miRNAs are highly stable in the TME and may serve as critical intercellular communication molecules, allowing cancer cells to manipulate or diminish surrounding recipient T cells through horizontal transfection [17]. miR-762 serves as an exosomal microRNA from OSCC cells (Supplementary Figure S1). Furthermore, we found that miR-762 not only suppressed NF-κB signaling transactivation (Figure 1D) but also E-cadherin promoter transcription activity (Figure 1E), which may inhibit immune cell maturation and prevent migration into the TME. These results suggest that miR-762 plays an important role in the OSCC-TME with immune suppression function. We speculate that miR-762 may lead to decreased T-cell activation and function in the OSCC-TME.
2.2. Exogenous miR-762 Inhibits the Jurkat Cells Activation and Migration Ability
To demonstrate the function of extracellular miR-762 in the OSCC-TME, particularly in tumor immune evasion through the prevention of T-cell activation, we transfected Jurkat cells with miR-762-PM for 72 h. The surface T-cell activation markers, CD25 and CD69, were inhibited by miR-762 transfection (Figure 2A). Additionally, the proinflammatory cytokine IL-12, which augments the growth, differentiation, and activation of cytotoxic CD8 T cells [18], was reduced by miR-762 manipulation compared to the NC control (Figure 2B). This indicates that miR-762 inhibited Jurkat-cell cytotoxic activity.
Furthermore, miR-762 transfection reduced Jurkat cell proliferation (Figure 2C) and Transwell migration ability (Figure 2D). These results show that miR-762 is an important suppressor of T-cell migration and activation in the TME. Additionally, we introduced miR-762 into Cal-27 cells, which have both lower intracellular and secretion levels of miR-762, and harvested the cell culture supernatant as an OSCC-derived conditioned medium to mimic the TME. The miR-762-containing Cal-27 conditioned medium (Cal-27-miR-762-CM) also suppressed PHA-activated Jurkat cell migration and further inhibited the gene expression of T-cell activation markers such as IL2RA, CD69, and CD71 (Figure 2F). Taken together, these results indicate that miR-762 in the TME can inhibit T-cell recruitment and activity.
2.3. Exogenous miR-762 Inhibits T-Cell CXCR3 Expression
CXCR3 is primarily expressed in T cells, dendritic cells, and natural killer cells, playing a crucial role in T-cell trafficking and function [19]. Moreover, CXCR3 has three variants, A and B, which exhibit differences in their expression profiles in the TME and have different functions in cancer progression [20]. TargetScan analysis predicted a direct binding interaction between miR-762 and CXCR3 (Figure 3A). We performed a CXCR3-3′-UTR reporter assay with or without the miR-762 binding site mutation. The CXCR3-3′-UTR was significantly suppressed by miR-762 mimic (PM) transfection but was abolished by the miR-762 binding site mutation (Figure 3B). Additionally, ectopic miR-762 expression directly suppressed CXCR3 mRNA and protein expression in Jurkat cells (Figure 3C–E). These results indicate that miR-762 could regulate CXCR3 through both reducing mRNA stability and protein translational inhibition. Moreover, miR-762 selectively reduced the expression of the T-cell proliferation and migration-promoting variant CXCR3A.
To further examine the role of extracellular miR-762 in the OSCC tumor microenvironment, we applied an OSCC-conditioned medium to Jurkat cells to assess CXCR3 expression. The conditioned medium harvested from miR-762-expressing Cal-27 cells also suppressed CXCR3 expression in Jurkat cells (Figure 3G,H). These results indicate that OSCC-secreted miR-762 can suppress T-cell CXCR3 expression in the oral tumor microenvironment. Both direct transfection of miR-762 and OSCC-derived miR-762 conditioned medium were found to stimulate the expression of the CXCR3B variant, which is generated through alternative splicing (Figure 3F–H). Unlike CXCR3B, which exhibits tumor-suppressive functions, CXCR3A promotes tumorigenesis by enhancing cell proliferation and migration. The regulation of CXCR3B involves unique transcription factors, NRF2 and BACH1 [21], which modulate its expression and downstream effects in response to miR-762. This suggests that miR-762 may influence the alternative splicing or transcriptional regulation of CXCR3 isoforms, thereby shaping their distinct roles in tumor progression and immune modulation. Additionally, CXCR3B functions as a receptor for CXCL4, a chemokine known to inhibit cytotoxic T-cell migration and activation [22]. These findings suggest that miR-762 may exert a unique T-cell suppression function through the CXCL4/CXCR3B axis, distinct from the tumor-promoting role of CXCR3A.
2.4. CXCR3 Promotes Cytotoxic T-Cell Activation
To further examine whether CXCR3 contributes to T-cell activation, we evaluated the T-cell activation status following ectopic CXCR3 expression (Figure 4A). We found that CXCR3 expression enhanced the expression of T-cell activation genes such as IL2RA (CD25), CD69, and CD71 mRNA (Figure 4B). Additionally, we observed that CXCR3 expression increased the expression of T-cell activation surface markers CD69, CD25, and CD154 in Jurkat cells (Figure 4C). Moreover, we also found that CXCR3 increased Jurkat T-cell proliferation (Figure 4D). IL-12, a T cell-stimulating cytokine, promotes the growth and function of T cells and the production of interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) from T cells and natural killer (NK) cells [23]. CXCR3 expression also stimulated IL-12 secretion from Jurkat cells (Figure 4E).
Taken together, our results show that CXCR3 enhances Jurkat T-cell cytotoxicity by promoting T-cell polarization and proliferation and increasing IL-12 secretion, thereby proving that CXCR3 plays a crucial role in T-cell activation.
2.5. miR-762/CXCR3 Suppression Axis Is Critical in Environment T-Cell Activation
The molecular mechanisms underlying CXCR3 regulation of T-cell function are not fully understood. Several studies have suggested that CXCR3 signaling involves Ras/ERK, Src, and PI3K/AKT pathways [24]. Additionally, CXCL10/CXCR3 activation can stimulate PI3K/AKT signaling, thereby promoting Th1 cell differentiation and migration. The PI3K/AKT pathways are then stimulated, leading to the promotion of cell survival, proliferation, and cytotoxic activity of memory T and NK cells.
To clarify the interplay among the miR-762/CXCR3/Akt axis in OSCC immune evasion, we investigated whether OSCC miR-762-induced CXCR3 downregulation plays a critical role in T-cell activation. To this end, we generated a CXCR3 expression plasmid lacking the 3′-UTR sequence, thereby preventing inhibition by miR-762. In the miR-762 mimic-transfected Jurkat cells, restoring CXCR3 expression (Figure 5A) could restore AKT activity (Figure 5B). The restored CXCR3 expression further reactivated T-cell polarization (Figure 5C,D) and proliferation ability (Figure 5E). Moreover, the critical T-cell cytokine IL-12 secretion was also restored by CXCR3 expression (Figure 5F).
In summary, these results suggest that the miR-762/CXCR3 axis may regulate T-cell activation, polarization, proliferation, and IL-12 secretion in the tumor microenvironment.
2.6. Horizontal Transmission of miR-762 Promotes Immune Escape in OSCC
Finally, we aimed to demonstrate the horizontal transmission of miR-762 between OSCC cells and T cells, which regulates T-cell cytotoxicity and promotes OSCC immune escape. To do this, we used an indirect coculture system (Figure 6A). When miR-762 was introduced into low-expressing Cal-27 cells, T-cell migration (Figure 6B) and cytotoxicity (Figure 6C) were abolished by miR-762 expression. Moreover, this cancer immune escape could be reversed by reintroducing CXCR3 into the T cells.
Taken together, these results suggest that miR-762 attenuates T-cell cytotoxicity against cancer by reducing the expression levels of CXCR3. These findings indicate that T-cell inactivation induced by OSCC is primarily mediated by OSCC-derived miR-762.
3. Discussion
OSCC employs various immune-escape mechanisms, primarily involving inhibitory lymphocytes and other immune cells that release cytokines suppressing cytotoxic CD8 lymphocytes. miR-762, an exosomal microRNA from mice epithelial cells [25]. This study identified that horizontal transmission of miR-762 from OSCC cells may contribute to immune escape by suppressing CXCR3 expression and activating the CXCR3-AKT signaling pathway in T lymphocytes. Chemokines in the TME play a crucial role in cancer progression by promoting tumorigenesis [26]. However, chemokines also recruit adaptive immune cells, enhancing anti-tumor immunity. For instance, CXCR3, a receptor for chemokines CXCL9, CXCL10, and CXCL11, is more highly expressed in tumor tissues than in normal tissues [27]. In cancer cells, autocrine CXCR3 signaling promotes metastasis and growth through AKT signaling [8], while CXCL9 and CXCL11 can inhibit tumor growth by facilitating immune cell infiltration [28]. Thus, tumors must find new ways to evade the immune system. Horizontal gene transfer (HGT) is vital in TME [29]. MicroRNAs HGT quickly inhibits recipient cells gene expression by being continuously supplied from cancer cells. MicroRNAs are easily neutralized by complementary miRNA sequences [30]. After COVID-19 pandemic, RNA-based medicines offer new therapeutic options for disease [31]. Our discovery of miR-762 in OSCC immune escape via horizontal transmission to immune cells in the TME suggests a new niche for RNA-based cancer therapies.
OSCC is typically an immune-cold tumor, showing lower T and NK cell infiltration [32]. CXCR3 is vital for lymphocyte migration and activation, and miR-762 in the TME may help maintain an immunosuppressive environment. In addition to aiding immune escape during tumor initiation, the immune cell content in TME is crucial for predicting treatment outcomes and patient prognosis [33]. miR-762 distribution in TME prevents immune cell infiltration and worsens responses to conventional cancer treatments. Using anti-sense miR-762 could remove miR-762 from the TME, enhance immune cell infiltration, and boost anti-tumor cytotoxicity, improving responses to chemotherapy and radiotherapy.
In OSCC, CXCR3 enhances lymphatic invasion and cancer growth [34], while CXCR3A promotes cancer stemness and chemoresistance [35], indicating CXCR3 is important for OSCC progression. In late-stage NSCLC, CXCR3 expression is diminished in both CD4 and CD8 T cells [36]. CXCR3 is highly expressed in adaptive immune cells, aiding in chemotaxis and activation. Reduced NK-cell CXCR3 expression hinders immune cell recruitment to tumors [37]. Suppressing CXCR3 can promote immune escape. Interestingly, miR-762-treated OSCC-CM increases CXCR3B expression, which acts as the CXCL4 receptor [38]. CXCL4/CXCR3B has opposite effects to CXCL10 in T cells, reducing proinflammatory IFN-gamma and increasing TH2 cytokines [39]. CXCL4 inhibits activated T-cell proliferation [40] and stimulates Treg growth [41]. CXCR3B expressions may enhance OSCC immune escape and immunosuppressive environments via miR-762 transmission. In summary, a novel OSCC escape mechanism through miR-762 transmission, preventing immune cell infiltration and anti-tumor cytotoxicity, may offer new niches for restoring anti-OSCC immunity.
4. Materials and Methods
4.1. Chemical Reagents, Kits, Antibodies, and Primers
All reagents, kits, antibodies, and primer sequences are listed in Table S1. CXCR3 vectors were created by Gateway cloning, as described in our previous study [42]. Lentiviral particles were generated by Taiwan RNAiCore standard protocol [43]. The target cells were infected and antibiotic selection. The miR-762-PM transfection mix was prepared using TransIT-X2 according to the manufacturer’s protocol and applied to Jurkat cells.
4.2. Cell Culture Conditions
Human acute T-cell leukemia Jurkat cells., human embryonic kidney 293T, and human oral cancer cells SCC-4, SCC-9, SCC-15, SCC-25, Cal-27, Cal-33, HSC-2, HSC-3, HSC-3-M3, and HSC-4, Ca9-22, OSC-19, OSC-20, and SAS were purchased from the original resource and listed in Table S1. All cells were cultured in the standard culture condition by following the instruction manual and maintained for 3 months. Additionally, all cells were regularly checked for mycoplasma infection and cell morphology to ensure cell health.
4.3. OSCC Conditioned Medium Preparation
Conditioned medium from OSCC was prepared using Cal-27 cells transfected with either miR-762-PM or control microRNA (Genomics, New Taipei City, Taiwan). Briefly, 50 nM of microRNA was transfected into Cal-27 cells using TransIT-X2 reagents (Mirus Bio, Madison, WI, USA). After 16 h of incubation, the transfected cells were washed and replaced with serum-free medium, followed by an additional 24 h incubation. The conditioned medium was then collected, filtered through a 0.22 μm PVDF filter to remove cell debris, and stored at −80 °C until further use. OSCC exosomes were harvested from ultra-centrifuge purification methods [44].
4.4. Activation of Jurkat T-Cell
The Jurkat cells were stimulated with 1.25 μg/mL of anti-CD3 (Clone: OKT3; BioLegend, San Diego, CA, USA) monoclonal antibody (mAb) and 1.25 μg/mL of anti-CD28 (Clone: CD28.2; BioLegend) mAb for 72 h. Jurkat-CXCR3 cells were stimulated with the same amount of anti-CD3 and anti-CD28 mAb plus 10 ng/mL of IL-12 (R&D Systems, Inc., Minneapolis, MN, USA) for 72 h.
4.5. T-Cell Cytotoxicity Assay Against OSCC Cells
In this study, 2 × 10^4^ Jurkat cells were seeded in the upper-Transwell chamber (Wuxi NEST Biotechnology, Wuxi, China) and co-transfected with or without miR-762 mimics and CXCR3-CDS expression vector for 72 h. For T-cell cytotoxicity, 1 × 10^4^ Cal-27 cells were seeded in the lower well. After 48 h, Cal-27 cells were fixed in 70% ice-cold ethanol for 60min, then stained with a 0.1% crystal violet/20% methanol solution and recorded under a light microscope.
4.6. Gene Expression Analysis
Total RNA was isolated by TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA). cDNA was synthesized by Roche First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland). All primers were listed in Table S1. qRT-PCR analysis was used with ChamQ Universal SYBR qPCR Master Mix, (Vazyme Biotech, Nanjing, China), Gunster Biotech MB-Q96-LP qPCR plate (Gunster Biotech, New Taipei City, Taiwan) and QuantStudio 3 qPCR system Thermo Fisher Scientific, Waltham, MA, USA). For miRNA detection, cDNA was prepared by the QIAGEN miScript II RT Kit (Qiagen, Hilden, Germany). Antibodies were listed in Table S1. Furthermore, 10–30 μg protein lysates were loaded onto % SDS-PAGE and blotted to PVDF membranes. The protein expression was analyzed by the e-BLOT Touch Imager (e-BLOT, Shanghai, China). Supernatants were collected and analyzed by IL-12 ELISA kits (R&D System, Minneapolis, MN, USA).
4.7. T-Cell Cytotoxicity and Migration Assays
In this study, 2 × 10^4^ Jurkat cells were seeded in the 0.4-μm upper chamber and co-transfected with miR-762-PM and CXCR3-CDS vector for 72 h. For the T-cell cytotoxicity assay, 1 × 10^4^ Cal-27 cells were seeded in the lower well. For migration assays, Jurkat cells were seeded in the 3 μm upper chamber and incubated with different lower chamber conditions for 24 h. The cytotoxicity of OSCC cells (48 h) or migrated Jurkat cells was fixed, stained, and recorded by microscope (200× magnification, Leica, Wetzlar, Germany). miR-762-PM-transfected Jurkat cells were seeded and incubated for 72 h by CCK-8 assay. Moreover, 10% CCK-8 (dojindo, Kumamoto, Japan) was added and incubated for an extra 3 h, then measured OD450 by Thermos Multiskan reader (Thermo Fisher Scientific, Waltham, MA, USA).
4.8. Reporter Assays for Transcription and 3′UTR Reporter Activity
For the NF-kB and CDH1 activity assay, the experiment was performed in Cal-27 cells. For the 3′-UTR reporter assays, the CXCR3 3′-UTR region was cloned by PCR amplification (446 bp after the CXCR3 3′-UTR stop codon) and cloned into the modified CpoI sites of the pCMV-Greenfire-3′-UTR vector (System Biosciences, Palo Alto, CA, USA). The pGF-CXCR3-UTR miR-762 binding site mutation clone was generated by site-directed mutagenesis with the primer listed in Supplementary Table S1. The CXCR3 3′UTR-reporter-expressed Cal-27 cells were seeded into 24-well plates and co-transfected with 25 nM miR-762 mimics or NC control. All CXCR3-UTR reporters were packaged into pseudovirus particles and introduced into Cal-27 cells. After 48 h, the 3′-UTR reporter activity was measured by the ONE-Glo™ substrate (Promega, Madison WI, USA) and SpectraMaxID3 reader (Molecular Device, San Jose, CA, USA).
4.9. Flow Cytometry Analysis
To determine surface markers, cells were labeled with the surface CD25 and CD69 for 60 min on ice in the dark. The stained cells were analyzed using an Attune NxT Flow Cytometer (Thermo Fisher Scientific, Waltham, MA, USA), and the quantitative analysis was performed using FlowJo v10 (Becton-Dickson, Franklin Lakes, NJ, USA)
4.10. Statistical Analysis
All experiments were repeated at least 3 times. Data were presented as mean ± standard deviation (SD). Differences between various treatment groups were assessed using Student’s t test. Between-group differences were considered significant at p < 0.05. Data analyses were performed using GraphPad Prism version 8 (Dotmatics, Boston, MA, USA).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Siegel R.L. Miller K.D. Wagle N.S. Jemal A. Cancer statistics, 2023 CA Cancer J. Clin.202373174810.3322/caac.2176336633525 · doi ↗ · pubmed ↗
- 2Chou C.W. Lin C.R. Chung Y.T. Tang C.S. Epidemiology of Oral Cancer in Taiwan: A Population-Based Cancer Registry Study Cancers 202315217510.3390/cancers 1507217537046836 PMC 10092957 · doi ↗ · pubmed ↗
- 3Elmusrati A. Wang J. Wang C.Y. Tumor microenvironment and immune evasion in head and neck squamous cell carcinoma Int. J. Oral Sci.2021132410.1038/s 41368-021-00131-734341329 PMC 8329257 · doi ↗ · pubmed ↗
- 4Gajewski T.F. Schreiber H. Fu Y.X. Innate and adaptive immune cells in the tumor microenvironment Nat. Immunol.2013141014102210.1038/ni.270324048123 PMC 4118725 · doi ↗ · pubmed ↗
- 5Kim S.K. Cho S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment Front. Pharmacol.20221386869510.3389/fphar.2022.86869535685630 PMC 9171538 · doi ↗ · pubmed ↗
- 6Hughes C.E. Nibbs R.J.B. A guide to chemokines and their receptors FEBS J.20182852944297110.1111/febs.1446629637711 PMC 6120486 · doi ↗ · pubmed ↗
- 7Kohli K. Pillarisetty V.G. Kim T.S. Key chemokines direct migration of immune cells in solid tumors Cancer Gene Ther.202229102110.1038/s 41417-021-00303-x 33603130 PMC 8761573 · doi ↗ · pubmed ↗
- 8Wang X. Zhang Y. Wang S. Ni H. Zhao P. Chen G. Xu B. Yuan L. The role of CXCR 3 and its ligands in cancer Front. Oncol.202212102268810.3389/fonc.2022.102268836479091 PMC 9720144 · doi ↗ · pubmed ↗