Phylogenetic Relationship and Characterization of the Complete Mitochondrial Genome of the Cuckoo Species Clamator coromandus (Aves: Cuculidae)
Yu Zhang, Hao Gao, Fan Zhang, Chengxing Xia, Guopan Li, Shaobin Li

TL;DR
This study provides the first complete mitochondrial genome of the chestnut-winged cuckoo and explores its evolutionary relationships within the Cuculidae family.
Contribution
The first complete mitochondrial genome of Clamator coromandus is sequenced and analyzed for phylogenetic and genetic insights.
Findings
The mitogenome is 17,082 bp with typical Cuculidae organization and high AT content in control regions.
Phylogenetic analysis shows C. coromandus is closer to Piaya cayana than Ceuthmochares aereus.
Brood parasitism and parental care species show distinct genetic differences and multiple evolutionary transitions.
Abstract
The chestnut-winged cuckoo (Clamator coromandus) is a bird species known for its brood parasitism, laying eggs in the nests of other bird species. However, there is a paucity of genetic information available for this species and their genus Clamator. In this study, we present the first complete mitochondrial genome sequence of C. coromandus and compare it with other species within the Cuculidae family. The mitogenome is a closed circular molecule consisting of 17,082 bp with an organization typical of the mitochondrial genomes of Cuculidae. Alignment of the control regions across Cuculidae species revealed substantial genetic variation and a significant abundance of AT content. A significant difference was detected in AT-skews between brood-parasitic and parental-care species. A distinctive long poly-C sequence was located at the 5′ end of domain I. Phylogenetically, C. coromandus is…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
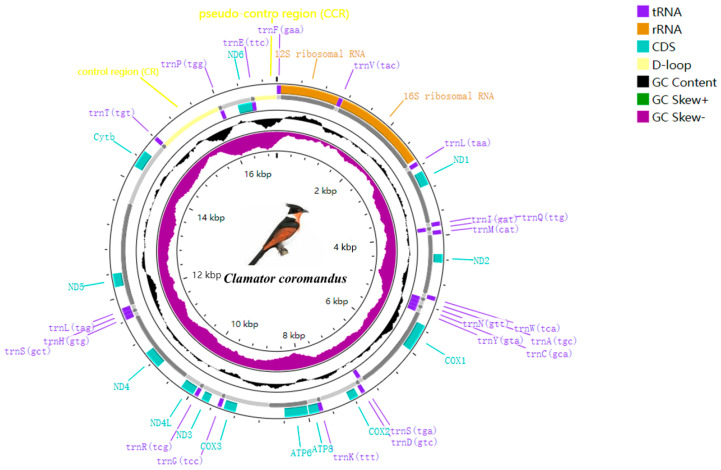 Figure 1
Figure 1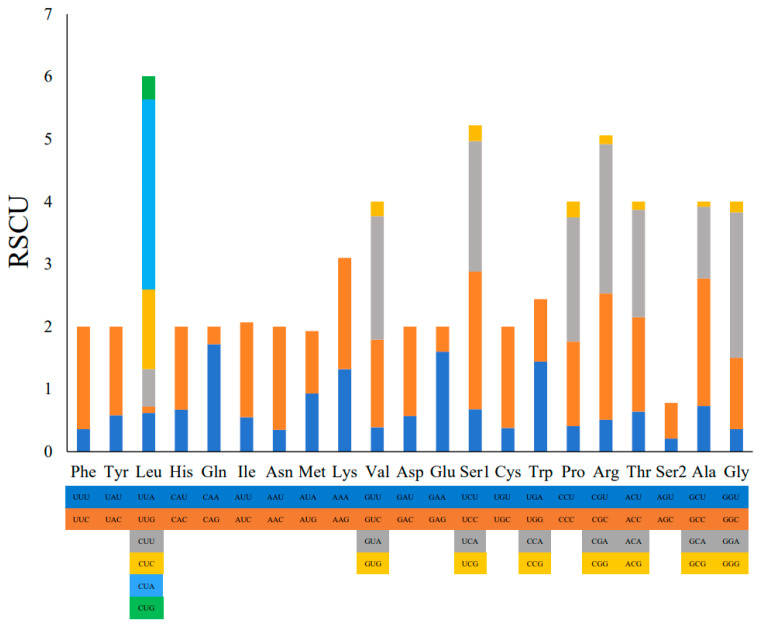 Figure 2
Figure 2 Figure 3
Figure 3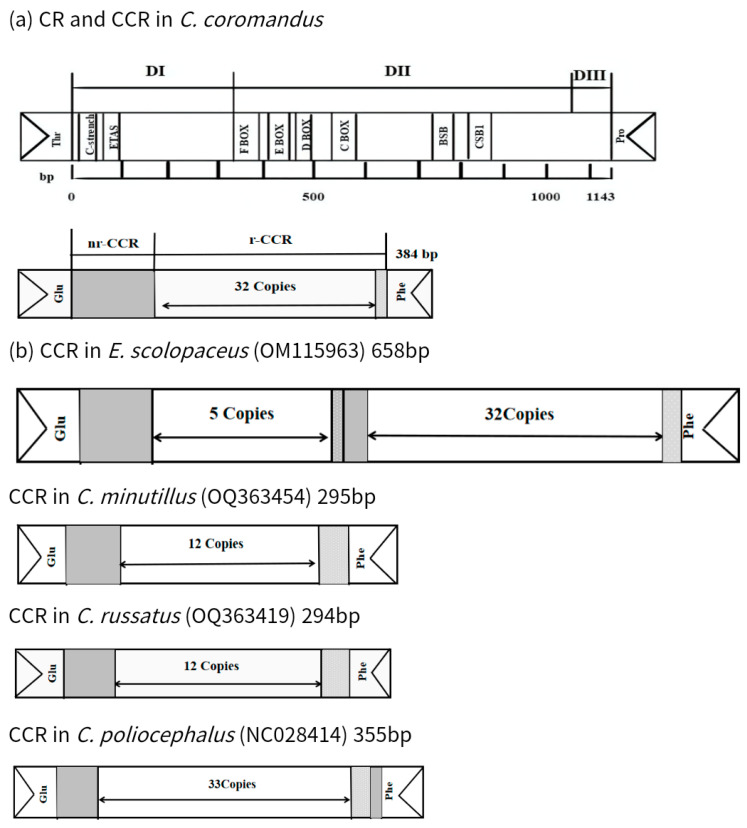 Figure 4
Figure 4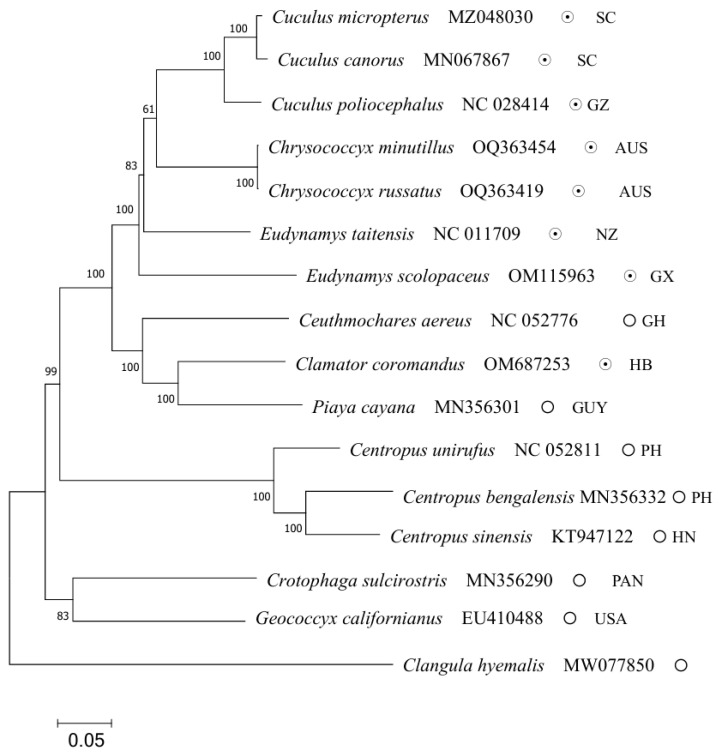 Figure 5
Figure 5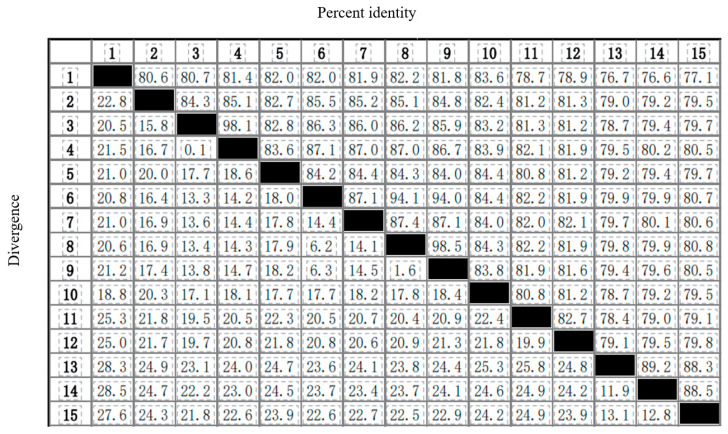 Figure 6
Figure 6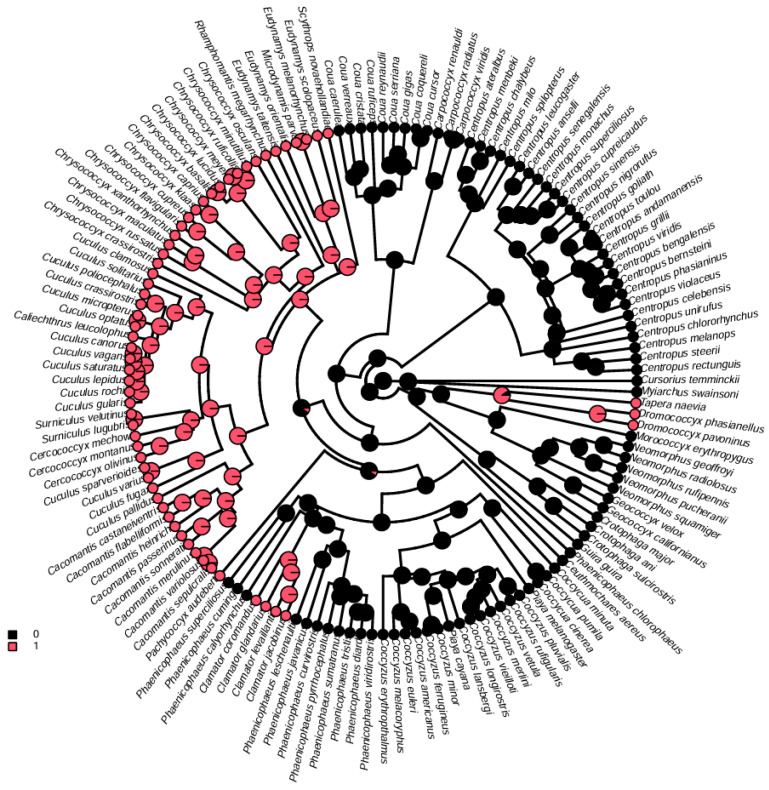 Figure 7
Figure 7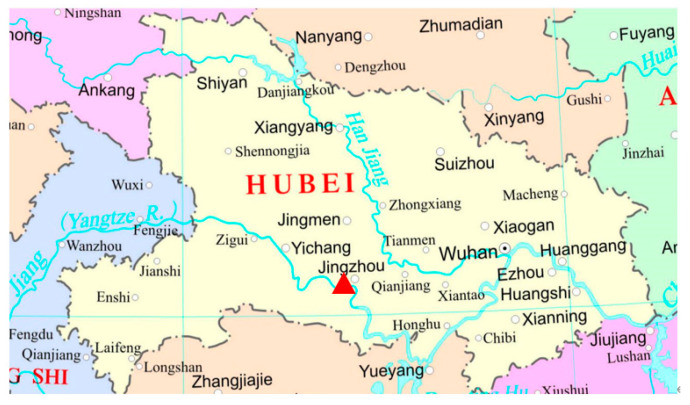 Figure 8
Figure 8- —National Natural Sciences Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Genetic diversity and population structure · Plant and Fungal Species Descriptions
1. Introduction
The family Cuculidae, the cuckoos, malkohas, ani’s, and roadrunners (147 species in 24 genera) [1], is notable for its diverse social behaviors and reproductive strategies. It is best known for its brood-parasitic members, which lay their eggs in the nests of other bird species, thereby relinquishing parental responsibilities [1,2]. Brood parasitism has evolved independently at least three times within this family, with many species exhibiting this behavior [3]. These cuckoos parasitize a remarkable variety of hosts globally, often laying eggs that closely mimic those of their hosts. Some species’ young may even eject host eggs from the nest upon hatching [4]. Primarily forest dwellers, these birds are often elusive, detected more by their distinctive, simple whistled calls than by sight.
The chestnut-winged cuckoo (Clamator coromandus), a member of the genus Clamator, is well-known for its striking plumage and unique behaviors [5]. The genus Clamator currently contains four species, but little information is available on them. The chestnut-winged cuckoo is a medium-sized bird, weighing between 66 and 86 g. It is notable for its metallic glossy black plumage, spiky crest, and narrow white nape-band. This species breeds across northern India and Nepal, extending eastward to southern and eastern China, with an altitudinal distribution up to 2450 m [6,7]. The chestnut-winged cuckoo typically lays its eggs in the nests of other bird species. As an insectivorous and migratory species, it winters in southern India, Sri Lanka, and the Greater Sundas. It thrives in diverse habitats, from wooded areas to scrub, bushes, orchards, plantations, and tall reedbeds [8].
However, C. coromandus remains a poorly understood species, with existing knowledge primarily limited to basic species accounts [1,4,5]. This study addresses this gap in research concerning its genetic characteristics by sequencing and reporting the complete mitochondrial genome of C. coromandus. We generated new data from this species and compared them with mitogenome sequences of other cuckoo species available in GenBank (Table 1). Our objectives were threefold: (a) to elucidate the sequences, features, and structures of the mitochondrial genome of C. coromandus, (b) to compare the mitogenome sequences between interspecific brood parasitism and parental care species, and (c) to describe the taxonomic status and phylogenetic relationships of C. coromandus within the family Cuculidae. The complete mitochondrial genome provides valuable insights into the evolutionary history and phylogenetic positioning of C. coromandus and related species within the broader avian phylogeny. Our findings contribute to a better understanding of species identification and genetic evolution within the Cuculidae family.
2. Results
2.1. Genome Content and Organization
The complete mitochondrial genome (mitogenome) of C. coromandus is a closed circular molecule and 17,082 bp in length. It contains the typical set of 37 genes found in vertebrate mitogenomes, including 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, 2 ribosomal RNA (rRNA) genes (12S rRNA and 16S rRNA), a control region (CR), and an additional pseudo-control region (CCR) (Table 2, Figure 1). Compared to the classical mitochondrial structure, the mitogenome of C. coromandus has four duplicate genes (ND6, trnT, trnP, and trnE) and an extra CR, referred to as a CCR. ND6 and 8 tRNAs are transcribed from the light strand (L), while the other 12 PCGs, 14 tRNAs, 2 rRNAs, and 2 noncoding regions (CR, CCR) are located on the heavy (H) strand. The nucleotide composition of the mitogenome of C. coromandus is biased towards A and T (55.87%: A = 32.46%, T = 23.42%, G = 13.10%, and C = 31.03%). In the full mitogenome of C. coromandus, the AT- and GC-skews were 0.16 and −0.41. The A + T content of the CR was notably higher than that of the PCGs, tRNAs, and rRNAs (Table S1).
Mitochondrial genes overlapped by a total of 35 bp across 11 different locations, ranging from 1 to 10 bp. The longest overlap (10 bp) was found between the ATP6 and ATP8 genes, while the shortest overlap (1 bp) was observed in 12S rRNA-tRNA-Phe, tRNA-Val-12S rRNA, 16S rRNA-tRNA-Val, tRNA-Leu-16S rRNA, tRNA-Met-tRNA-Gln, COXⅢ-ATP6, and tRNA-Leu-tRNA-Ser genes. Additionally, some PCGsshared 1–10 nucleotides with adjacent tRNA genes. Furthermore, 17 intergenic spacers were present in the mitogenome of C. coromandus, totaling 86 bp. The longest spacer sequence was 15 nucleotides long, located between the ND1 and tRNA-Ile genes (Table 2).
AT/GC skews were essentially similar in the mitochondrial PCGs, two rRNA genes, and the mitogenomes of other species of Cuculidae, except for a few genes (ND1, ND3, and ND6; Table S2). Notably, Centropus bengalensis exhibited a negative AT-skew in, while the other fourteen species had positive AT-skews (Table S2). For ND3 genes, Centropus unirufus showed a negative AT-skew, whereas the other fourteen species had positive AT-skews. Apparent positive AT-skew and negative GC-skew biases were identified for all 12 PCGs (except for ND1, ND3, and ND6) encoded by the H-strand, whereas the reverse was detected in ND6 encoded by the L-strand (Table S2).
From the perspective of overall mitochondria and 13PCGs (Table S2), both brood parasites and parental care species exhibited similar AT/GC skews, with positive AT-skews and negative in GC-skews. Nonparametric tests (Table 3) revealed significant differences between brood parasites and parental care species in the AT-skews of overall mitochondria and 13PCGs (p < 0.05), while no significant differences were found in GC-skews (p > 0.05).
2.2. Protein-Coding Genes and Codon Usage Patterns
The 13 mitochondrial PCGs of C. coromandus span 11,391 bp, which account for 66.68% of the total mitogenome sequence. These PCGs consist of 3797 codons in total. Most PCGs use ATG as the start codon, except for ND3 and ND6, which start with ATT and CTA, respectively (Table 2). TAA is the most frequent stop codon in the mitogenome of C. coromandus. COXⅢ, ND4, and ND6 have incomplete stop codons (T-), ND1 and ND5 end with AGA, ND2 ends with TAG, and COXI ends with AGG. All other stop codons are the standard terminal codon (TAA). Among the 64 available codons, the most commonly used ones were for proline (P; CCA; 3.92%), threonine (T; ACA; 3.95%), phenylalanine (F; UUC; 4.19%), isoleucine (I; AUC; 5.38%), and leucine (L; CUA; 7.31%). Leucine was the most frequently used amino acid in C. coromandus (Table 4, Figure 2). In contrast, the codons UUG, GUG, GCG, UGU, CGU, CGG, and GGG were rarely used, accounting for only 1.14% of the amino acids. Among the 60 codons analyzed, 28 had Relative Synonymous Codon Usage (RSCU) values greater than or equal to 1.00, indicating a preference for these codons. Leucine (Leu) has a higher preference for CUA, and the RSCU value is the highest in mitochondrial genome PCGs, which is 3.03. Methionine (AUG) and Tryptophan (UGG) showed no codon preference (RSCU = 1) (Table 4). The stop codons UAA, UAG, AGA, and AGG do not encode amino acids. Most amino acids have at least two different codons, while leucine has six codons (Figure 2). The base composition of the three codon positions of the 13 PCGs is shown in Table S3. The A + T content is 51.89% at the first position, 58.78% at the second position, and 54.54% at the third position. Hence, the second position has the highest A + T content, whereas the first position has the highest C + G content (48.11%; second position: 41.22%; third position: 45.46%). Moreover, the first positions and third positions have a positive AT-skew and negative GC-skews, but the second positions have negative AT- and GC-skews (Table S3). Among the 13 PCGs in the mitogenome of C. coromandus (Table 2), ATP8 has the highest A + T content (58.9%), and COXI has the lowest (52.9%).
2.3. Transfer and Ribosomal RNA Genes
The mitogenome of C. coromandus contains the typical set of 22 tRNA genes with conventional secondary structures. The total length of mitochondrial tRNA genes is 1542 bp, with individual genes sizes ranging from 65 bp (tRNA-Cys) to 74 bp (tRNA-Leu1 and tRNA-Ser1). Most tRNAs can be folded into the canonical cloverleaf secondary structure, except for tRNA-Ser (GCT), which lacks the DHU arm and loop but contains the TΨC arm and loop. A total of 31 unmatched base pairs were found in the tRNAs of C. coromandus, with the most common being G-U (22 instances), followed by U-U (2 instances), A-C (4 instances), U-C (1 instance), A-A (1 instance), and C-C (1 instance) (Figure S1). The 12S rRNA gene is located between tRNA-Phe and tRNA-Val, while the 16S rRNA gene is situated between tRNA-Val and tRNA-Leu (Figure 1). The lengths of the 12S rRNA and 16S rRNA were 975 bp and 1596 bp, respectively (Table 2). The base composition of the 12S rRNA gene is 20.21% T, 26.97% C, 32.92% A, and 19.9% G, with an A + T content of 53.13%. The 16S rRNA gene has a base composition of 21.74% T, 25.31% C, 34.71% A, and 18.23% G, with an A + T content of 56.45%.
2.4. Noncoding Regions
The noncoding region of the mitogenome of C. coromandus comprises two major control regions. The first CR is 1143 bp long and is located between tRNA-Thr and tRNA-Pro. The second is a short noncoding region, referred to as the CCR, which is 384 bp in length and situated after the tRNA-Glu gene (Figure 1). The CCR sequence is the shortest among all sequences in the mitogenome (Table 5). The base frequencies in the CR of C. coromandus are 30.53% T, 27.12% C, 29.31% A, and 13.04% G, whereas those in the CCR are 12.50% T, 31.25% C, 54.95% A, and 1.30% G.
Hypervariable domain I (D I), located to the left (5′ end) of the CR, typically contains tandem repeat sequences with a copy number ranging from 1 to 8. The copy number varies both between species and among individuals within a species. At the 5′-end of D I, we observed a long continuous poly-C sequence (Figure 3), which appears to be a conserved feature. Conserved palindromic motifs 5′-TACAT-3′ and 5′-ATGTA-3′ were identified at the 5′ end of the CR of C. coromandus (Figure 3). The four conserved boxes (C, D, E, and F) and the CSB1 regions were also found in C. coromandus (Figure 3 and Figure 4a).
As shown in Figure 4a, the CCR of C. coromandus contains a 157 bp nonrepeating region (nr-CCR) at the 5′ end, followed by a cluster of tandem repeats at the 3′ end (r-CCR): 32 units of 7 bp repeat units (followed by a 6 bp incomplete repeat unit) (Figure 4, Table 5). In Eudynamys scolopaceu, two clusters of repetitive regions were observed in the CCR (Figure 4b): the first cluster (66 bp per unit, 5 times) at the 5′ region, and the second cluster (7 bp per unit, 23 times) at the 3′ region. These two regions are separated by a 28 bp nr-CCR and show no similarity to each other.
2.5. Phylogenetic Relationships
To explore the molecular phylogenetics and evolution of Cuculidae, a phylogenetic analysis was conducted based on the nucleotide sequence data of the 13 PCGs of C. coromandus and 14 other species ofCuculidae. Clangula hyemalis was used as the outgroup. The maximum likelihood tree is shown in Figure 5. The maximum likelihood trees showed stable topological structures with high nodal support values. Based on 14 other complete mitogenome sequences retrieved from NCBI GenBank (Table S1), most internal nodes were well-supported by bootstrap values (Figure 5). The results indicate that the genus Eudynamys is not monophyletic because Eudynamys taitensis was more closely related to species of Cuculus and Chrysococcyx than to E. scolopaceus (Figure 5). The phylogenetic tree reveals that among the 14 other species of Cuculidae, C. coromandus was closely related to Ceuthmochares aereus and Piaya cayana, with C. coromandus being most closely related to P. cayana. Comparisons of genetic divergence also indicated that C. coromandus was most similar P. cayana (Figure 6). The phylogenetic tree also indicates a general separation between interspecific brood parasitism and parental care, with a transition towards brood parasitism occurring twice within the evolutionary history of the cuckoos, once in the lineage leading to C. coromandus and once in the lineage leading to the clade formed by Eudynamys, Cuculus, and Chrysococcyx. We have marked the locations of the sampling points on the phylogenetic tree and found that all sampling sites of species in the Cuculus genus are located in China, with all species within this genus being brood parasites (Figure 5). From a phylogenetic perspective, cuckoos, as migratory birds, are distributed across various regions worldwide. We reconstructed the ancestral characteristics of the Cuculidae family (Figure 7). The phylogenetic analysis indicates a transition from parental care to brood parasitism within the cuckoo lineage. The results of our ancestral state reconstruction align with those presented in Figure 5.
3. Discussion
The complete mitochondrial genome of C. coromandus is a closed circular molecule, which is similar to other members of the Cuculidae family. It contains the typical set of 37 genes found in vertebrate mitogenomes. This configuration is consistent with other Cuculidae species [9]. Previous studies have suggested that the extra CR could confer selective advantages, such as a replicative advantage, to duplicated mitochondrial DNAs over those with a single CR [10,11]. In addition, we observed mitochondrial genome rearrangements in C. coromandus. Specifically, an additional noncoding sequence was found between the tRNA-Glu and tRNA-Phe genes aside from the original control region located between tRNA-Thr and tRNA-Pro [12]. The presence of the CCR is noted in the mitochondrial genomes of numerous bird species [13]. Other Cuculiformes species also possess a CR and a short CCR [14,15]. Similar rearrangements have been detected in representatives of Passeriformes, Procellariiformes, Falconiformes, Piciformes, and Psittaciformes [16,17,18]. Studies have also found that the duplication of CRs in the mitochondrial genome is associated with longer lifespans in birds compared to mammals of similar weight [19]. The gene arrangement pattern of the mitochondrial genome of C. coromandus is identical to that of other species of Cuculidae, all of which have the remnant CCR gene order, differing from the standard bird gene order [20,21]. AT-skew, GC-skew, and A + T content are commonly used to assess differences in the nucleotide composition of mitochondrial genomes [22]. This mitogenome has greater A + T content (55.87%) (Table S1) compared to the G + C content (44.13%), which is similar to other species of Cuculidae [23,24]. Some PCGsshare nucleotides with adjacent tRNA genes. This compact and economical arrangement is a common feature of mitochondrial DNA [25].
COXⅢ, ND4, and ND6 have incomplete stop codons (T-). These incomplete stop codons are presumably completed through post-transcriptional polyadenylation using a poly-A tail [26]. Codon preference refers to the phenomenon where specific codons are utilized more prevalently than others in the DNA or RNA sequences of certain organisms, which can influence gene expression and protein assembly [27]. Leucine (Leu) has a higher preference for CUA. Methionine (AUG) and Tryptophan (UGG) showed no codon preference. Consistent with other vertebrates [28], there is a strong bias against G at the third codon position in all 13 PCGs. The G content is 5.93% at the third codon position (Table S2).
Meanwhile, tRNA-Ser (GCT) lacks a DHU arm and loop. This feature is typical of vertebrate mitogenomes [29] and is generally observed in vertebrate tRNA genes [30]. Structural compensation mechanisms may render the missing arm in tRNA-Ser functional [31]. Most anticodons are identical to those observed in other Cuculidae species, and the CCA 3′-terminal group is added during processing. A total of 31 unmatched base pairs were found in the tRNAs of C. coromandus. Stem mismatches are common in tRNA genes and are likely repaired through post-transcriptional editing [32]. Compared to other birds, most mismatched nucleotides were G–U pairs, which can form weak bonds in tRNAs and noncanonical pairs in tRNA secondary structures [33]. The gene order, arrangement, length, base composition, and RNA structure of the ribosomal RNA genes (12S rRNA and 16S rRNA) are similar to those in other Cuculidae birds.
Both vertebrates and invertebrates CRs exhibit high A + T content and possess replication initiation features [34]. The A + T content of the CR of C. coromandus is 59.8%, and the CCR is 67.5% (Table 2). The control region of C. coromandus shares structural similarities with Chrysococcyx minutillus and Chrysococcyx russatus, and it exhibits common characteristics found in other birds [35]. Generally, three distinct CR domains are recognized: (a) the highly variable, left-end domain I (D I); (b) the conserved, central domain (D II); and (c) the right-end domain III (D III) [36]. A conserved palindromic motif can be identified at the 5′ end of the CR of C. coromandus [37]. The CCR of C. coromandus contains a cluster of tandem repeats (Figure 4, Table 5). In E. scolopaceu, two clusters of repetitive regions were observed in the CCR (Figure 4b). These variable tandem repeats are the primary cause of length variability in mitochondrial genome control regions and the entire mitogenome [38,39].
In this study, a phylogenetic analysis was conducted on the nucleotide sequence data of 13 PCGs from C. coromandus and 14 otherspecies of Cuculidae. C. hyemalis was selected as the outgroup. The complete mitochondrial genome of C. coromandus will hopefully contribute to the systematic and molecular identification of Cuculidae. The phylogenetic trees indicate that E. scolopaceus is not a sister-taxon of E. taitensis. Our current study provides further evidence for a closer phylogenetic relationship between C. coromandus and P. cayana compared to other species included in our data [40], suggesting the need for further investigation. C. coromandus, as a brood-parasitic species, is more closely related to parental care (P. cayana) (Figure 5). Species that are more similar in terms of brood parasitic behavior are not necessarily more closely related in the phylogeny of the Cuculidae family [41]. This result is consistent with our study. All clades were well-resolved, illustrating that despite their rapid evolutionary rates, mitogenomes possess species-specific evolutionary relationships that can be effectively elucidated through enhanced taxon sampling [42]. By adding sampling points for species on the phylogenetic tree, we observed that cuckoos inhabit numerous regions globally; however, all sampling points for species within the Cuculus genus are confined to China. Research into the migration routes and wintering grounds of Cuculus canorus has demonstrated that their migration paths exhibit relative stability with minimal variation, ultimately leading them to winter in Africa [43,44]. From the perspective of the phylogenetic tree, the sampling point for C. canorus was situated in Sichuan Province, China, thereby providing valuable data for studying the migratory behavior of this species. Darwin initially proposed that parasitic cuckoos evolved from parental cuckoos [45]. Our findings demonstrate a general distinction between interspecific brood parasitism and parental care, with transitions between these behaviors occurring at least twice within the evolutionary history of cuckoos. Following our reconstruction of ancestral traits within Cuculidae (Figure 7), we found that these results corroborate findings from this study, which supports the hypothesis regarding a transition from parental care to interspecific brood parasitism.
4. Materials and Methods
4.1. Sample Collection and Genomic DNA Extraction
Muscle tissue from a male C. coromandus (Figure S2) was collected from an individual that died accidentally from glass collision near the east gate of the ancient city in Jingzhou County, Hubei Province, China (latitude: 30°20′5″ N; longitude: 112°12′57″ E; Figure 8) on 29 October 2022. This species can be easily distinguished by its body size and plumage patterns [46]. The sex of the individuals was determined using a pair of universal primers for avian sex identification [47]. Whole-genome DNA was extracted from the muscle tissue according to the protocol of TIANamp Genomic DNA kits (Tiangen, Beijing, China). The remaining sample is now stored in herbarium room 317 of the #1 Teaching Building on the West campus of Yangtze University (www.yangtzeu.edu.cn, Dr. Shaobin Li, [email protected]).
4.2. Mitochondrial DNA Amplification and Sequencing
High-throughput sequencing technology was employed to sequence the mitochondrial genome of C. coromandus from the muscle tissue. Genomic DNA was extracted, and a DNA library was constructed using the TruSeq Nano DNA HT Sample Prep Kit (Illumina, San Diego, CA, USA). The library was sequenced on an Illumina HiSeq/NovaSeq platform with a 2 × 150 paired-end sequencing strategy.
4.3. Assembly, Annotation, and Analysis of the Mitochondrial Genome
The complete mitochondrial genome sequence of C. coromandus was assembled using the SeqMan module of DNASTAR software (DNASTAR, Inc., Madison, WI, USA) [48]. PCGs were identified through sequence comparisons with known sequences of other birds using the CLUSTAL W program. Transfer RNA (tRNA) genes and their secondary structures were identified using tRNAscan-SE 1.21 [49] or based on their proposed secondary structures and anticodons [50]. Ribosomal RNA genes (rRNAs) were identified via NCBI BLAST search and comparisons. An online tool, CGVIEW, was employed to draw the mitogenome structure map (https://cgview.ca/ accessed on 17 January 2025) [51]. Codon usage and nucleotide composition statistics were computed using MEGA 7.0 [52]. If RSCU = 1, there is no preference for the use of this codon, and if RSCU > 1, the codon is used preferentially by amino acids, while if RSCU < 1, the codon usage is contrary. Composition skew analysis was performed with the formulas AT-skew = [A − T]/[A + T] and GC-skew = [G − C]/[G + C] [53]. The 15 species of Cuculidae were divided into two categories, namely, brood parasites and parental care (Table 1). We employed the non-parametric Mann–Whitney U test to determine whether AT/GC-skew nucleotides differ between brood parasites and parental care using SPSS 20.0 software. Because the sample size was too small (brood parasites: n = 8; parental care: n = 7) and the two groups of data did not follow a normal distribution, non-parametric tests were used for comparison. Based on the next-generation sequencing method, the complete sequence of the mitochondrial genome of C. coromandus was assembled and compared with the species of the same family reported in GenBank to determine the start and end points of the control region. By conducting a comprehensive alignment of the reported mitochondrial control region structures in birds [35,54,55,56], we analyzed the structure of the mitochondrial control region of C. coromandus and identified three regions within its control region: the termination-associated sequence (ETAS), the central conserved region (CD), and the conserved sequence block (CSB). The core of ETAS is TACAT, and its reverse complementary sequence is ATGTA, which can form a hairpin structure. In the CD, three characteristic fragments were identified, namely CSB-D, CSB-E, and CSB-F. The CSB typically contains three conserved sequences: CSB1, CSB2, and CSB3. The sequence of the complete mitogenome of C. coromandus was deposited in GenBank under accession number OM687253.
4.4. Phylogenetic Analysis
The PCGs were extracted from mitogenomes using PhyloSuite v.1.1.16 [57]. Repetitive sequences were removed to obtain a final set of protein-coding regions for constructing the phylogenetic tree. Sequence data were initially aligned using ClustalX 1.83 [58] with default parameters. The mitogenome of C. coromandus was analyzed in this study, along with 14 other Cuculidae mitogenomes retrieved from NCBI GenBank (Table S1). The concatenated sequences of the 13 PCGs of complete mitochondrial genomes were used. The best-fit substitution models (GTR + G + I) of nucleotide sequences were tested using MEGA. We evaluated the confidence of each branch by performing 1000 bootstrap replications. Finally, the trimmed PCGs were concatenated to construct a phylogenetic tree using MEGA. We added the sampling points of species samples on the phylogenetic tree. With the exception of the sampling site for C. coromandus, all other sampling locations for 14 additional cuckoo species were obtained from previous studies [14,15,23,24,59,60]. We download 1000 alternative phylogenetic trees of cuckoos from birdtree.org [61] and constructed a maximum clade credibility tree, which is adequate for addressing phylogenetic uncertainty [62]. The parasitic behavior state of ancestral nodes is considered the response variable, while the parasitic behavior of extant cuckoo species (i.e., non-brood parasitic cuckoos or brood parasitic cuckoos) is considered the independent variable (n = 142 species). We reconstructed ancestral states of brood-parasitism’s presence using the “ace” Function in R package, and our model choice was “ER” (equal-rates model), with the type being discrete character [63]. All 142 species of cuckoos included in the analysis are present in previous studies [3,4,6,41,64]. All analyses were carried out in R 4.3.3 [65] using the R package phytools [66].
5. Conclusions
This study represents the first comprehensive report and analysis of the complete mitochondrial genome of C. coromandus. The mitogenome structure of C. coromandus is typical for birds and shows high similarity to other reported Cuculidae mitogenomes. Our results revealed a significant difference in AT-skews between interspecific brood parasitism and parental care. Notably, a long continuous poly-C sequence was discovered at the 5′ end of the displacement loop (D-loop) region. Additionally, one type of tandem repeat unit was identified in the CCR. Phylogenetic analyses indicate a closer relationship between C. coromandus and P. cayana compared to C. coromandus and C. aereus. While interspecific brood parasitism and parental care are generally separated, transitions between these behaviors occur occasionally within the evolutionary tree. This suggests that both traits have evolved multiple times across different evolutionary lineages. Consequently, our findings provide a valuable resource for future studies on the mitochondrial genomic evolution of the Cuculidae family. However, many aspects of Cuculidae phylogeny remain unresolved. Further analysis using additional molecular markers, such as nuclear genes or the genome, and larger taxon sampling is necessary to clarify the phylogenetic relationships among species within this family.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Billerman S.M. Keeney B.K. Rodewald P.G. Schulenberg T.S. Birds of the World Cornell Laboratory of Ornithology Ithaca, NY, USA 2022 Database
- 2Wang N. Shan C. Chen D. Hu Y. Sun Y. Wang Y. Liang B. Liang W. “Isolation by Gentes with Asymmetric Migration” shapes the genetic structure of the common cuckoo in China Integr. Zool.20252014415910.1111/1749-4877.1285338872343 · doi ↗ · pubmed ↗
- 3Hasegawa M. Arai E. Differential visual ornamentation between brood parasitic and parental cuckoos J. Evol. Biol.20183144645610.1111/jeb.1324029336511 · doi ↗ · pubmed ↗
- 4Erritzøe J. Mann C.F. Brammer F. Fuller R.A. Cuckoos of the World Christopher Helm London, UK 2012
- 5del Hoyo J. Elliott A. Christie D. Handbook of the Birds of the World, Vol. 10: Cuckoo-Shrikes to Thrushes Dickinson E.C. Dekker R. Lynx Edicions Barcelona, Spain 2005
- 6Yang C. Liang W. Antonov A. Cai Y. Stokke B.G. Fossøy F. Moksnes A. Røskaft E. Diversity of parasitic cuckoos and their hosts in China Chin. Birds 2012393210.5122/cbirds.2012.0004 · doi ↗
- 7Zheng G.M. A Checklist on the Classification and Distribution of the Birds of China Science Press Beijing, China 2023
- 8Wu J. Shi Y. Attribution index for changes in migratory bird distributions: The role of climate change over the past 50 years in China Ecol. Inform.20163114715510.1016/j.ecoinf.2015.11.013 · doi ↗