Tnni3k regulates cardiomyopathy and cardiac conduction disease through Nfatc1 signaling
Shi Ouyang, Yujuan Niu, Le Liu, Qiaorong Yi, Wuming Qin, Hui Cao, Tao Liao, Rong Xiang, Yonghe Ding, Yun Deng

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
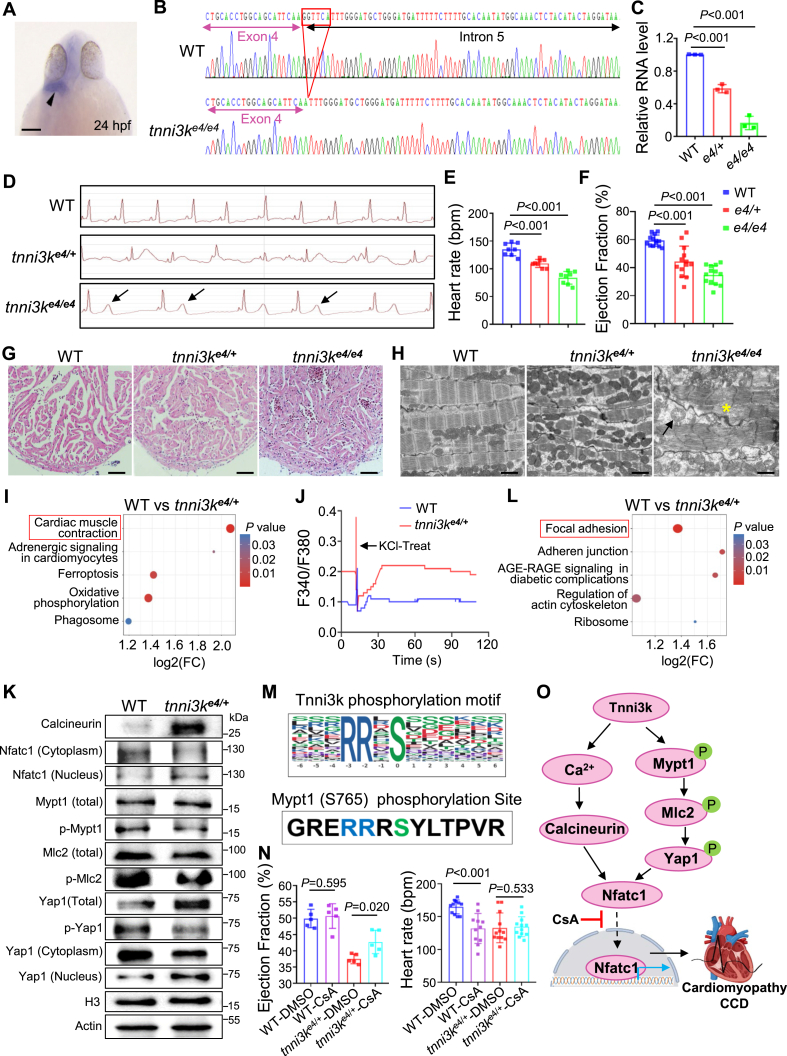 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac Fibrosis and Remodeling · Cardiomyopathy and Myosin Studies · Ubiquitin and proteasome pathways
TNNI3K (troponin-I interacting kinase) encodes a duo tyrosine and serine/threonine kinase implicated in cardiomyopathy, arrhythmias, and cardiac conduction disease (CCD).1 However, no direct downstream phosphorylation targets of TNNI3K have been identified yet.2 Here, we employed the CRISPR/Cas9 gene-editing technique to generate a splicing mutation in the 4th exon of zebrafish tnni3k ortholog gene that mimics a TNNI3K splicing variant identified from a patient family with cardiomyopathy and CCD. This tnni3k heterozygous mutant (tnni3k^e4/+^) recapitulated several key features of cardiomyopathy and CCD, thus representing a novel model of human TNNI3K mutation-based cardiac diseases. Next, we utilized this heterozygous tnni3k^e4/+^ mutant to perform proteomics and phosphoproteomic analysis. As a result, we identified Mypt1/Mlc2/Yap1/Nfatc1 axis as the downstream phosphorylation targets of Tnni3k. Lastly, we found that treatment of cyclosporine A, an inhibitor of Nfatc1 translocation from cytoplasm to the nucleus, exhibited a partial cardioprotective effect on the tnni3k^e4/+^ heterozygous mutant. Together, we generated a unique zebrafish animal model of TNNI3K-based cardiac diseases and identified the Mypt1/Mlc2/Yap1/Nfatc1 axis as its downstream phosphorylation targets that could be potentially leveraged to develop new therapeutic strategies.
The zebrafish tnni3k gene exhibits a cardiac-specific expression pattern and encodes a protein that shares approximately 85 % amino acid identity with human TNNI3K (Fig. 1A; Fig. S1), supporting TNNI3K as an evolutionally highly conserved protein across species. A previous study reported that a novel splice site mutation in the 4th exon of the human TNNI3K gene (c.333 + 2T > C) caused dilated cardiomyopathy and CCD.3 To mimic the cardiomyopathy and CCD caused by this human TNNI3K variant, we designed a single guide RNA targeting the splicing donor site of the 4th exon in the zebrafish tnni3k gene and generated a splicing mutant in the predicted splice donor site via CRISPR/Cas9 technology (Fig. 1B; Fig. S2A–C). This splicing mutation resulted in aberrant tnni3k splicing between exon 4 and exon 5 and led to a premature stop of Tnni3k protein translation. As a result, the mRNA level of tnni3k was significantly reduced in both the tnni3k heterozygous (tnni3k^e4/+^) and homozygous (tnni3k^e4/e4^) mutants compared with that in the wild-type sibling, likely due to a nonsense-mediated mRNA decay mechanism (Fig. 1C). Next, we carried out detailed phenotypic analysis on these tnni3k splicing mutants. First, we found T wave elevation in the tnni3k homozygous but not heterozygous mutant (Fig. 1D). Second, we detected significant cardiac function decline and heart rate reduction in both the tnni3k^e4/+^ and tnni3k^e4/e4^ mutants (Fig. 1E, F). Third, we found obvious increased myocardium muscle density, sarcomere degeneration, mitochondrial swelling, and/or Z-disc distortion phenotypes, most notably in the tnni3k^e4/e4^ homozygous mutant hearts (Fig. 1G, H). Collectively, we generated a splice site mutation in the zebrafish tnni3k gene that led to cardiac dysfunction and conduction disorder, recapitulating several key features of human TNNI3K mutation-caused cardiomyopathy and CCD.Figure 1. Tnni3k regulates cardiomyopathy and cardiac conduction disease through Nfatc1 signaling. (A) Whole-mount in situ hybridization (WISH) assay for detecting endogenous expression patterns of the tnni3k gene at 24 h post-fertilization (hpf). The black arrows point to the WISH signal in the heart. Scale bar, 100 μm. (B) The chromographs illustrating the sequences of the wild-type (WT) tnni3k gene and the mutant alleles with a 6-base pair (bp) nucleotide deletion (red box) in the splicing site adjacent between the 4th exon and 5th intron at the genomic levels. (C) The tnni3k transcript levels between WT and mutant alleles detected by quantitative real-time PCR. n = 3, one-way Analysis of Variance (ANOVA). (D, E) Representative electrocardiogram (ECG) recordings (D) and heart rate quantification analysis (E) in the tnni3k mutants and WT sibling controls at 6 months. The arrows indicate T-wave elevation. n = 8, one-way ANOVA. bpm, beats per minute. (F) Ejection fraction (EF) (in %) measured by echocardiography in the indicated tnni3k mutant lines and WT sibling controls at 6 months. n = 8–13, one-way ANOVA. (G) Representative hematoxylin & eosin (H&E) staining of the apex area of the ventricle in the tnni3k mutants and WT sibling controls at 6 months. Scale bars, 100 μm. (H) The confirmative transmission electron microscopy (TEM) images showing mitochondrial swelling/empty (arrow) and myofibril degeneration phenotype (asterisk) in the tnni3k^e4/e4^ homozygous, but not obvious in the tnni3k^e4/+^ heterozygous mutant hearts at 6 months. Scale bars, 5 μm. (I) The top five enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with significant fold changes in expression levels and P values resulting from proteomic analysis between the tnni3k^e4/+^ mutants and WT sibling controls. The cardiac muscle contraction pathway was affected most significantly, and proteins in this pathway were subsequently experimentally tested. (J) Increased [Ca^2+^]i was detected in the tnni3k^e4/+^ mutant cardiomyocytes compared with WT controls. (K) Representative western blotting images on the indicated protein expression levels in the tnni3k^e4/+^ mutant hearts compared with the WT sibling controls at 6 months. (L) The top five enriched KEGG pathways with significant fold changes in expression levels and P values resulting from phosphoproteomic analysis between the tnni3k^e4/+^ mutants and WT sibling controls. The focal adhesion pathway was highlighted, and proteins in this pathway were focused on with subsequent experimental validation. (M) Putative Tnni3k phosphorylation substrate motif was predicted using MoMo (http://meme-suite.org/tools/momo). (N) Percent ejection fraction measured by echocardiography and heart rate measured by ECG in the tnni3k^e4/+^ mutant fish compared with the WT sibling controls at 6 months with or without cyclosporine A (CsA) treatment. n = 5–10, one-way ANOVA. (O) Proposed working model. Tnni3k functions by regulating calcineurin/Nfatc1-mediated calcium homeostasis and phosphorylating members of the Mypt1/Mlc2/Yap1/Nfatc1 axis to maintain cardiac contraction and heart rhythm. CCD, cardiac conduction disease.Figure 1
To identify downstream protein targets of Tnni3k phosphorylation and gain insights into the pathways regulated by the Tnni3k protein underlying the molecular mechanisms of aberrant tnni3k splicing-induced cardiomyopathy and CCD, we performed both quantitative proteomic and phosphoproteomic analysis using protein lysate isolated from the tnni3k^e4/+^ heterozygous mutant hearts, as the mimicking TNNI3K splice site variation identified from the human patient is a heterozygous mutation.
First, through the proteomic analysis, we identified 212 differentially expressed proteins in the tnni3k^e4/+^ hearts compared with the wild-type controls based on a fold change >1.3 and a P value < 0.05 (Fig. S3B). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that the most prominently affected pathway was cardiac muscle contraction (Fig. 1I). As studies have suggested that cardiac muscle contraction is regulated mainly by the intracellular free Ca^2+^ concentration ([Ca^2+^]i),4 we then focused on its downstream cascade reaction of the Ca^2+^/calcineurin/Nfatc1 signaling pathway for further experimental validation. We examined [Ca^2+^]i by imaging the intracellular calcium concentration and detected an increase of [Ca^2+^]i in the tnni3k^e4/+^ splicing mutant cardiomyocytes compared with the wild-type controls (Fig. 1J). We then evaluated the total protein levels of the calcineurin and Nfatc1 proteins, which are two positive downstream effectors of Ca^2+^. Our results showed that calcineurin and nuclear Nfatc1 were significantly up-regulated, while cytoplasmic Nfatc1 (cytoplasm) was down-regulated in the tnni3k^e4/+^ mutant hearts compared with the controls (Fig. 1K; Fig. S3E). Thus, these proteomic analysis results indicated that Tnni3k might function by regulating cardiac muscle contraction and/or Ca^2+^/calcineurin/Nfatc1 homeostasis to maintain normal cardiac function and rhythm.
Second, through the phosphoproteomic analysis, we found that protein phosphorylation levels were increased at 102 sites of 83 proteins and decreased at 518 sites of 310 proteins. KEGG enrichment of the differentially expressed phosphorylated proteins showed that focal adhesion was among the most significantly affected pathways in the tnni3k^e4/+^ mutant hearts (Fig. 1L). We further performed the phosphorylation motif analysis which predicted that the Tnni3k target proteins carried arginine-arginine-X-serine amino acid residues (Fig. 1M). Interestingly, among the top differentially expressed phosphorylated protein targets identified as being associated with focal adhesion, Mypt1 carried the same phosphorylation site (S765) as the predicted targeting motif of the Tnni3k protein. Thus, we performed a more detailed experimental validation analysis on the Mypt1 protein and its downstream effectors, such as Mlc2 and Yap. Indeed, we confirmed that phosphorylated Mypt1 and Yap1 proteins were down-regulated significantly in the tnni3k^e4/+^ hearts compared with the wild-type controls, and subsequently, nuclear Yap1 were significantly up-regulated, while cytoplasmic Yap1 (cytoplasm) was down-regulated in the tnni3k^e4/+^ hearts (Fig. 1L; Fig. S3E), consistent with the data from the proteomic and phosphoproteomic analyses. Taken together, this phosphoproteomic analysis identified the Mypt1/Ml2/Yap1 axis as the likely downstream phosphorylation target of the Tnni3k protein.
Next, considering that Mypt1/Yap1 is an upstream effector of Nfatc1, regulating the translocation of Nfatc1 into the nucleus,5 we tested the hypothesis that pharmacologic blockade of Nfatc1 into the nucleus in the tnni3k mutant would confer a cardioprotective effect on the tnni3k splicing mutation-based cardiomyopathy and CCD. To do so, we treated the tnni3k^e4/+^ mutant and wild-type sibling control fish with cyclosporine A, an inhibitor of Nfatc1 in the nucleus, which acts by restraining the activity of Ca^2+^-dependent calcineurin. The results showed that cyclosporine A treatment indeed led to a statistically decreased level of protein in the nucleus in both the tnni3k^e4/+^ mutant and wild-type control hearts (Fig. S4). More importantly, cyclosporine A treatment partially restored the cardiac function decline, but seemed to have no impact on the heart rate reduction in the tnni3k^e4/+^ mutant (Fig. 1N). Thus, these data indicated that pharmacologic inhibition of Nfatc1 translocation from the cytoplasm into the nucleus through cyclosporine A treatment could exert a partial therapeutic effect on tnni3k splicing mutation-based cardiomyopathy.
In conclusion, we generated a unique zebrafish animal model of TNNI3K splicing mutation-based cardiomyopathy and CCD and identified the focal adhesion pathway and Mypt1/Mlc2/Yap1/Nfatc1 axis as the downstream phosphorylation targets of the Tnni3k protein. We found that pharmacological inhibition of the translocation of Nfatc1 into the nucleus partially alleviated the cardiomyopathy phenotypes in the heterozygous tnni3k splicing mutant zebrafish model (Fig. 1O). More studies are needed to validate these findings in large mammalian animal models and to develop NFATC1 nuclear translocation-targeted therapeutic strategies for TNNI3K gene mutation-based cardiomyopathy and CCD.
Author contributions
Shi Ouyang: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. Yujuan Niu: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – review & editing. Le Liu: Data curation, Methodology. Qiaorong Yi: Data curation, Methodology. Wuming Qin: Data curation, Formal analysis, Methodology. Hui Cao: Data curation, Methodology. Tao Liao: Data curation, Methodology. Rong Xiang: Data curation, Formal analysis, Validation. Yonghe Ding: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. Yun Deng: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Ethics declaration
The animal study protocols were approved by the Institutional Animal Care and Use Committees of Hunan Normal University (No. 2019003) and Qingdao University (No. QDU-AEC-2023201).
Funding
This study was supported in part by the 10.13039/501100001809National Natural Science Foundation of China (No. 31970504, 31772548, 82371863, 82070394), the NHC Key Laboratory of Birth Defect for Research and Prevention (Hunan Provincial Maternal and Child Health Care Hospital) (No. KF2021003), and the Postgraduate Scientific Innovation Fund of Hunan Province, China (No. CX2018B302).
Conflict of interests
The authors declared no conflict of interests.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Theis J.L.Zimmermann M.T.Larsen B.T.TNNI 3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy Hum Mol Genet 23212014579358042492531710.1093/hmg/ddu 297PMC 4189907 · doi ↗ · pubmed ↗
- 2Pham C.Muñoz-Martín N.Lodder E.M.The diverse roles of TNNI 3K in cardiac disease and potential for treatment Int J Mol Sci 2212202164223420397410.3390/ijms 22126422 PMC 8232738 · doi ↗ · pubmed ↗
- 3Fan L.L.Huang H.Jin J.Y.Whole exome sequencing identifies a novel mutation (c.333 + 2T > C) of TNNI 3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease Gene 648201863672935568110.1016/j.gene.2018.01.055 · doi ↗ · pubmed ↗
- 4Eisner D.A.Caldwell J.L.Kistamás K.Trafford A.W.Calcium and excitation-contraction coupling in the heart Circ Res 121220171811952868462310.1161/CIRCRESAHA.117.310230 PMC 5497788 · doi ↗ · pubmed ↗
- 5Meng K.P.Majedi F.S.Thauland T.J.Butte M.J.Mechanosensing through YAP controls T cell activation and metabolism J Exp Med 21782020 e 2020005310.1084/jem.20200053 PMC 739816332484502 · doi ↗ · pubmed ↗