Identification of Genetic Variants in Status Epilepticus Associated With Fever
Hiroaki Hanafusa, Hiroshi Yamaguchi, Naoya Morisada, Ming Juan Ye, Shizuka Oikawa, Shoichi Tokumoto, Masahiro Nishiyama, Kandai Nozu, Hiroaki Nagase

TL;DR
This study finds that genetic variants are less common in children with fever-related seizures (SEF) compared to those with developmental and epileptic encephalopathy (DEE).
Contribution
The study is the first to compare genetic variant detection rates between SEF and DEE.
Findings
Genetic variants were detected in 26.7% of SEF cases and 63.0% of DEE cases.
SEF cases only showed SCN1A variants, while DEE cases involved 16 different genes.
SEF and DEE may have distinct mechanisms for seizure onset.
Abstract
Status epilepticus associated with fever (SEF) is often encountered in pediatric emergency departments, and some patients develop neurological emergencies, such as acute encephalopathy (AE). Although numerous genetic variants of developmental and epileptic encephalopathy (DEE) have been reported, the frequency of these disease‐associated variants of SEF is unknown. The first aim of this study was to investigate the associated genetic variants of SEF. The second aim was to compare the variations in genes between SEF and DEE. This retrospective, clinical observational study included patients with SEF or DEE who visited Kobe University Hospital or Kobe University affiliated hospitals and provided consent for a genetic diagnosis of SEF or DEE between January 1, 2021, and December 31, 2022. Fifteen patients with SEF and 27 patients with DEE consented to a genetic diagnosis and were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
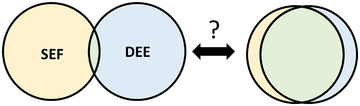 FIGURE 1
FIGURE 1| Patient no. | Sex | Age at onset | Diagnosis | Etiology | Seizure type | Seizure duration (min) | Prognosis (PCPC) | Birth history | Developmental history | Past medical history | Family history |
Brain imaging findings | Age at genetic test | Treatment at onset |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | M | 1y10m | SEF | Unknown | Tonic | 158 |
1 (prior to admission) 2 (at discharge) 1 (1m) 1 (6m) 1(1y) |
GA: 39w2d BW: 2886 g HT: 152 cm HC: 32.5 cm | No developmental delay in onset | None |
Autism (brother) |
CT (day 1): normal MRI (day10): normal | 1y11m |
DZPsp DZPiv fPHTiv PBiv TTM+BCT |
| Patient 2 | F | 1y11m | SEF | Parainfluenza3 | GTCS | 60 |
1 (prior to admission) 1 (at discharge) 1 (6m), 1 (1y) | GA: 41w0d BW: 2560 g | Generalized developmental delay, start to walk (1y9m) | None |
FS (grandfather, grandmother) |
CT (day1): subdural hematoma in the right parietal legion MRI (day8): right parietal, temporal and occipital hyperintensity (T1WI) | 2y |
MDLiv fPHTiv TTM+BCT |
| Patient 3 | M | 3m | AE (HSES) | Unknown | Tonic | 149 |
1 (prior to admission) 4 (at discharge) 4 (6m) 4 (1y) 4 (2y) | GA: 40w0d BW: 2900 g | No developmental delay in onset | None | None |
MRI (day14): cerebellar hemispheres and cerebral hemispheres show globally hypointensity (T1WI), hyperintensity (T2WI), hypointensity (FLAIR). Hyperintensity in the cortex and subcortical white matter, including the occipital and parietal lesions, and bilateral basal ganglia (DWI). Hyperintensity suggesting hemorrhagic necrosis in bilateral parietal and occipital gyri (T1WI) | 5y |
MDLiv, MDLcont PBiv SPT intubation |
| Patient 4 | F | 1y2m | SEF | Unknown | FBTCS | 231 |
1 (prior to admission) 2 (at discharge) 2 (1m), 3 (1y) | GA: 38w5d BW: 3062 g cesarean section because of breech position | Mild verbal developmental delay (1y3m), ESID: DQ78 (2y2m) | Afebrile seizures after vaccination (4m) | None | MRI (day9): normal | 1y3m |
MDLiv fPHTiv PBiv Thiiv TTM+BCT |
| Patient 5 | F | 2y7m | SEF | Adenovirus (rapid antigen test) | GTCS | 340 |
2 (prior to admission) 2 (at discharge) 2 (1m) 3 (6m) 3 (1y) 3 (2y) | GA: 37w1d BW: 2861 g | No developmental delay in onset |
Afebrile seizure (FIAS)(3m), FS (4m,5m,6m) | Developmental delay (brother) |
CT (day1): normal, MRI (day10): normal | 2y7m |
MDLom DZPsp DZPiv fPHTiv PBiv TTM+BCT |
| Patient 6 | F | 5y11m | SEF | Unknown | Tonic | 165 |
1 (prior to admission) 1 (at discharge) 1 (1m) 1 (6m) 1 (1y) | GA: 40w4d BW: 3495 g cesarean section | No developmental delay in onset | Afebrile seizure (4m), recurrent FSs | FS (mother) | CT (day1): normal, MRI (day8): normal | 6y |
DZPiv, fPHTiv PBiv TTM+BCT |
| Patient 7 | F | 8m | SEF | Unknown | FBTCS | 45 |
1 (prior to admission) 1 (at discharge) 1 (1m) | GA: 40w0d BW: 3620 g | No developmental delay in onset | Unilateral afebrile seizures (6m), FS (8m) | None | MRI (day13): mild degree of cerebral atrophy | 8y | MDLdiv TTM+BCT |
| Patient 8 | M | 1y0m | SEF | Unknown | FBTCS | 30 |
1 (prior to admission) 1 (at discharge) 1 (6m) 1 (1y) | GA: 38w0d BW: 2788 g | No developmental delay in onset | Microcephaly, Hypertrophic pyloric stenosis (2m), recurrent FS | Hyperthyroidism (mother) | None | 1y5m | DZPsp |
| Patient 9 | M | 4y0m | AE (HSES) | Unknown | GTCS | 120 |
2 (prior to admission) 5 (at discharge) 5 (6m) 5 (1y) 5 (2y) | GA: 37w0d BW: 2610 g | Language delay (2y4m), generalized delay (3y) | Recurrent FS | None | MRI (day2): hyperintensity throughout the cerebrum (DWI) | 6y | None |
| Patient 10 | F | 3y8m | AE (AESD) | Unknown | GTCS | 171 | 3 (prior to admission), 4 (at discharge) | GA: 37w3d BW: 2114 g | Without significant words, up to sitting position (3y) | Small jaw, early neonatal hypotonia, hypothyroidism | None | MRI (day6): BTA | 6y |
MDLiv Thiiv TTM+BCT, SPT |
| Patient 11 | M | 3y6m | AE (AESD) | Unknown | GTCS | 30 |
2 (prior to admission), 2 (at diacharge), 2 (1m) 2 (3m) 2 (6m) 2 (1y) 2 (2y) | GA: 37w5d BW: 2569 g | No significant words (3y) | Developmental delay | None | MRI (day10): BTA | 14y | DZPsp |
| Patient 12 | M | 1y5m | SEF | Unknown | GTCS | 249 |
1 (prior to admission), 1 (at discharge), 1 (1m) 1 (3m) 1 (6m) | Unknown | No developmental delay in onset | None | None | CT (day1): normal, MRI (day8): normal | 1y5m |
DZPiv MDLiv fPHTiv mitochondria rescue SPT TTM+BCT |
| Patient 13 | M | 9m | SEF | HHV6 (cerebrospinal fluid) | GTCS | 64 | 1 (prior to admission), 1 (at discharge), 1 (2m) | GA: 38w3d BW: 2698 g | Developmental delay (borderline) | Developmental delay | None | CT (day1): normal, MRI (day11): normal | 9m | MDLiv DZPiv, MDLcont, TTM+BCT |
| Patient 14 | M | 1y2m | AE (AESD) | HHV6 | GTCS | 90 | 1 (prior to admission), 4 (at discharge), 4 (6m) | GA: 38w4d BW: 2805 g | No developmental delay in onset | None | None |
CT (day1): global brain edema, MRI (day7): BTA, MRI (3m): Mild to moderate atrophy of the entire cerebrum | 1y3m |
DZPiv fPHT, MDLcont, SPT |
| Patient 15 | M | 1y6m | SEF |
Adenovirus, rhinovirus/ enterovirus, coronaHKU1, parainfluenza3 | GTCS | 81 | 1 (prior to admission), 1 (at discharge), 1 (6m) | GA: 39w5d BW: 3356 g | No developmental delay in onset | None | FS (father&mother) | CT (day1): normal, MRI (day8): normal | 1y8m |
MDLiv fPHTiv Thiiv, TTM+BCT |
|
Patient no. | Sex |
Age of onset |
Epileptic syndrome |
Seizure types |
Additional clinical findings |
Brain imaging findings | Birth history |
Developmental history | Family history |
Age at genetic test |
Anti‐epileptic medicine at genetic test | Other comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 16 | M | 2m | — | FBTCS |
Mild global developmental delay, Facial abnormality: nasal ridge, full cheek, eyebrows that are thin on the inside, brachycephaly, long philtrum |
MRI (2m): hypointensity of changes after subependymal hemorrhage (T2WI) and similar hypointensity in left occipital lobe subcortex |
GA: 37w6d BW: 2222 g (−1.73 SD, 4.2%tile) HT: 42 cm (−2.68 SD, 0.4%tile) HC: 31.7 cm (−0.94 SD,17.5%tile), asymmetrical SGA, APS: 3/5/8 | ESID: DQ55 at 7m | None | 10m | VPA, LEV | |
| Patient 17 | M | 5m | IESS | ES |
Macrocephaly, slight intellectual disability | MRI (5m): normal |
GA: 39w2d BW: 3780 g | Head control (3m), rolling over (4m), KSPD 2001: DQ88 at 1y5m |
Autism (niece), epilepsy&cerebral palsy (nephew) | 10m | VitB6, VPA |
EEG: hypsarrhythmia at seizure onset |
| Patient 18 | F | 1m | — |
Tonic FIAS GMS |
Autism, severe intellectual disability, insomnia | MRI (4m): normal |
GA: 40w0d BW: 3535 g | KSPD 2001: DQ22 at 5y8m | None | 15y | VPA, CZP | |
| Patient 19 | F | 8y | — | Myoclonic astatic |
Mild global developmental delay |
MRI (40y): cerebellar atrophy |
GA: 41w0d BW: 3050 g |
Head control (4m), start to walk (13m), slight developmental delay was pointed out at a previous hospital doctor at 8y, MMSE‐28 (40y), HDS‐R 26 (40y), FAB 16 (40y) | None | 40y | LTG, TPM, VPA, CLB, PER | |
| Patient 20 | F | 3m | — | Tonic | Severe intellectual disability | N/A |
GA: 38w2d BW: 2674 g |
Head control (8m), roll over (10m), no significant word (16y), developmental milestone was at the level of 8 months (16y) | None | 17y | VPA, LTG | |
| Patient 21 | F | 1d | — |
GTCS tonic clonic GMS | Severe intellectual disability, scoliosis | CT (4d): normal |
GA: 41w0d BW: 2548 g (IUGR), APS: 9/9 | KSPD 2001: DQ19 at 7y7m | None | 12y | PB, ZNS | EEG: suppression burst pattern at seizure onset |
| Patient 22 | M | 13m | — |
Tonic, myoclonic, FIAS | Severe intellectual disability |
MRI (15m): periventricular white matter hyperintensity (T2WI), MRI (2y): atrophy of frontal, temporal, and parietal lobes, MRI (8y8m): thinning of the entire cerebrum |
GA: 40w1d BW: 3354 g | Psychomotor developmental delay was noted from 8 months. No acquisition of development of sitting. No word and pursuit, bedridden (17y) | None | 17y | VPA, CLB | |
| Patient 23 | M | 1d | — |
Tonic FIAS | Severe intellectual disability |
CT (day2): normal, MRI (day4): normal |
GA: 40w0d BW: 2992 g | No head control, opisthotonus, no smiling at 1y3m | None | 1y3m | CBZ, PB | |
| Patient 24 | F | 2m | — |
FIAS FBTCS ES | At onset, generalized muscle weakness and severe generalized developmental delay, cafe au lait spots, axillary freckle‐like pigmented spots | MRI (2y2m): normal |
GA: 38w0d BW: 2684 g APS: 9/10 |
Head control (2y), pursuiting (1y10m), roll over (2y), sitting (2y8m), no significant word&no walking (3y) | None | 3m | VPA, PB, KBr, VitB6 | |
| Patient 25 | F | 6m | IESS | ES | Severe generalized developmental delay | MRI (1y8m): normal |
GA: 38w6d BW: 3500 g |
Developmental delay was pointed out at the 4‐month checkup, turning over and sitting (1y8m), stand up (3y4m), walking (3y6m), no word (3y6m), KSPD: DQ33 (1y7m) | None | 2y | VPA | |
| Patient 26 | F | 3m | — | Tonic | Severe intellectual disability | MRI (4m): normal |
GA: 41w6d BW: 3290 g APS: 9/10 |
Psychomotor developmental delay was pointed out at the 4‐month checkup. KSPD: DQ33 at 1y10m | None | 1y0m | CBZ, LEV | |
| Patient 27 | F | 6m | — |
GTCSS FIAS | Acquired microcephaly (45 cm at 3y, 47 cm 5y), stereotypic movements (hand kneading motion) | Unknown | Unknown |
Sitting (1y2m), the reaction to the surroundings has slowed down and the function of the fingers has decreased (1y5m) | None | 17y | LEV, CZP | |
| Patient 28 | M | 7y | — | FIAS | Mild developmental delay |
MRI (3y): arachnoid cyst, Chiari malformation type I |
GA: 37w4d BW: 3646 g |
Head control (3m), sitting (5m), standing up (10m), walking (1y5m), significant word(11m), KPSD: DQ70 (6y) | None | 8y | LEV | |
| Patient 29 | M | 3y | — | FIAS atonic | Mild developmental delay | CT (3y5m): mild dilation of left Sylvian fissure, arachnoid cyst |
GA: 38w6d BW: 2718 g | KSPD 2001: DQ68 (4y) | Epilepsy (Father) | 5y | TPM. VPA | |
| Patient 30 | M | 6m | — | GTCS | Mild verbal developmental delay, afebrile status epilepticus (3 times) | MRI (5m): normal |
GA: 39w2d BW: 2886 g |
Head control (4m), rolling over (6m), sitting (8m), standing up (10m), walking (1y8m), ESID: DQ63 (1y9m) | None | 7m | VPA, LEV | |
| Patient 31 | M | 6y | — | Myoclonic astatic | Severe intellectual disability, autism | CT (6y): normal, MRI (7y): normal |
GA: 40w0d BW: 2600 g cesarean section because of decreased heart rate |
Hypotonia, inability to walk and no word were noted at the health checkup (1y6m). Can walk but no speech (7y), KSPD 2001: DQ17 (6y) | None | 7y | VPA, PER | |
| Patient 32 | F | 6y | — |
FIAS tonic | Moderate intellectual disability | MRI (1y6m): normal |
GA: 38w0d BW: 2938 g |
Head control (4m), walking (1y3m), no word (22y) | None | 22y | VPA, RFM | |
| Patient 33 | F | 8m | — | Tonic | Moderate intellectual Disability | MRI (1y): normal |
GA: 38w0d BW: 2675 g |
Head control (4m), rolling over (5m), walking (1y), KSPD 2001: DQ45 (12y) | None | 1y1m | ZNS | |
| Patient 34 | F | 9m | — | FMS | Global developmental delay | MRI (1y1m): normal |
GA: 39w3d BW: 3242 g HT: 47.5 cm HC: 33.5 cm |
Head control (4m), walking (1y7m), no word (1y7m) | None | 1y1m | VPA, TPM, PER | |
| Patient 35 | M | 5m | IESS | ES | Moderate intellectual disability | MRI (6m): normal |
GA: 40w6d BW: 3900 g HC: 35 cm |
Rolling over (7m), no sitting (10m), ESID: DQ69 (6m), DQ77 (8.5m) | None | 6m | VPA | |
| Patient 36 | M | 12m | — |
ES GTCS tonic | Severe intellectual disability | N/A |
GA: 40w2d BW: 2800 g |
Head control (3m), sitting (7m), walking (1y6m), a few word (25y) | None | 25y | VPA, LTG, RFM | |
| Patient 37 | M | 2m | — | Tonic | Slight global developmental delay | MRI (2m): normal |
GA: 40w0d BW: 3634 g, HT:49.0 cm HC: 34.5 cm, Emergency cesarean section for stoppage of labor |
No sitting (9m), ESID: DQ80 (9m) | None | 2m | LCM | |
| Patient 38 | M | 13m | — | Absence | Mild global developmental delay, HPP, elevated urinary PEA | MRI (6m): normal |
GA: 37w5d BW: 2936 g, planned cesarean section |
Head control (5m), no sitting (9m), KPSI: DQ60 (1y) | Epilepsy (mother, two elder brothers, four aunts) | 1y5m | VitB6 | |
| Patient 39 | F | 6y | — |
GTCS FIAS | Mild global developmental delay | MRI (7y, 10y): normal |
GA: 38w0d BW: 2532 g APS: 9/10 | developmental delay was pointed out at a medical checkup (3y) KSPD: DQ54 (10y) | None | 14y | CLB, CBZ | |
| Patient 40 | M | 2d |
EIDEE & IESS |
FIAS tonic ES | Global developmental delay | MRI (2m): normal |
GA: 41w3d BW: 3736 g APS: 8/10 |
no pursuiting (4m), head control (5m), no rolling over, sitting and no word (9m) | None | 3m | VGB, LEV, VPA | |
| Patient 41 | F | 0d | — | FIAS | Severe intellectual disability | MRI (3y9m): frontal lobe atrophy | GA: 39w5d, BW: 2655 g, HC: 31.2 cm, APS: 9/10 |
sitting (6y), holdable (12y), no word (12y), KSPD: DQ13 (3y) | None | 12y | LTG | |
| Patient 42 | M | 3y | — | FIAS myoclonic | Mild intellectual disability | MRI (15y): normal | GA: 41w0d, BW: 3100 g, APS: 10/10 | KSPD: DQ44 (15y) | None | 15y | VPA, ESM, RFM, CZP, LEV |
| Patient No. | Identified method | gene | Variant | rs number | Inheritance | Zygosity | gnomAD frequency | ACMG guideline |
HGMD pro 2022.4 (number of references) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Patient 4 | Panel sequencing |
| De novo | Hetero | 0 | Pathogenic (PVS1+PS2+PM2) | DM(1) | ||||
| Patient 5 | Commercial |
| rs794726710 | De novo | Hetero | 0 | Pathogenic (PVS1+PS2+PM2) | DM(13) | |||
| Patient 7 | Commercial |
| NM_00165963.3:c.4398C>A,p.(F1466L) | Unknown | Hetero | 0 |
VUS (PM2+PM5 +PP3) | Novel | |||
| Patient 9 | WES |
| De novo | Hetero | 0 | Likely pathogenic (PS2+PM2) | Novel | ||||
|
| |||||||||||
| Patient 16 | Microarray |
| arr[GRCh37]6p21.33p21.32(30578599×2,30630793_32430238×1,32460307×2) | Unknown | Hetero | N/A | N/A | N/A | |||
| Patient 17 | Panel sequencing |
| NM_00314.4:c.737C>T,p.(P246R) | rs587782350 | De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | DM(16) | ||
| Patient 18 | Panel sequencing |
| rs61753251 | De novo | Hetero | 0 | Pathogenic (PVS1+PS2+PM2) | DM(12) | |||
| Patient 20 | Panel sequencing |
| rs587784453 | De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | DM(3) | |||
| Patient 23 | Panel sequencing |
| rs1057516098 | De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | DM(5) | |||
| Patient 24 | Panel sequencing |
| De novo | Hetero | 0 | Pathogenic (PVS1a+PS2+PM2) | Novel | ||||
| Patient 25 | Panel sequencing |
| De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | Novel | ||||
| Patient 26 | WES |
| De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | Novel | ||||
| Patient 27 | WES |
| De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | Novel | ||||
| Patient 28 | Panel sequencing |
| rs587776932 | De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | DM(13) | |||
| Patient 30 | Commercial |
| Unknown | Hetero | 0 | VUS (PM2+PP3) | DM?(4) | ||||
| Patient 31 | WES |
| De novo | Hetero | 0 | Pathogenic (PVS1+PS2+PM2) | Novel | ||||
| Patient 35 | Panel sequencing |
| De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | Novel | ||||
| Patient 37 | Panel sequencing |
| rs398123009 | De novo | Hetero | 0.000006572 | Pathogenic (PS2+PS3+PP3) | DM(42) | |||
| Patient 38 | Panel sequencing |
| rs387906525 | From mother | Hetero | 0.00001314 | Likely pathogenic (PVS1+PM2) | DM(20) | |||
| Patient 40 | Panel sequencing |
| De novo | Hetero | 0 | Pathogenic (PVS1+PS2+PM2) | Novel | ||||
| Patient 42 | Panel sequencing |
| rs797045599 | De novo | Hetero | 0 | Likely pathogenic (PS2+PM2+PP3) | DM(5) | |||
| SEF ( | DEE ( |
| |
|---|---|---|---|
| Original panel sequencing | 13 | 24 | 1.00 |
| Positive (%) | 1 (7.7) | 12 (50.0) | — |
| Whole exome sequence | 3 | 12 | 0.18 |
| Positive (%) | 1 (33.3) | 3 (25.0) | — |
| Microarray | 2 | 6 | 0.69 |
| Positive (%) | 0 (0) | 1 (16.7) | — |
| Others | |||
| Direct sequence | 2 | 2 | 0.61 |
| Commercial‐based genetic testing | |||
| Positive (%) | 2 (100) | 1 (50.0) | — |
- —Grant‐in‐Aid for Scientific Research
- —Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInfectious Encephalopathies and Encephalitis · Epilepsy research and treatment · Bacterial Infections and Vaccines
Introduction
1
Febrile seizure (FS) is the most common form of infection‐triggered seizures among children, affecting 2%–5% of those aged between 6 months and 6 years (Nishiyama et al. 2020; Subcommittee on Febrile Seizures, American Academy of Pediatrics 2011). Status epilepticus associated with fever (SEF) is often encountered in the pediatric emergency department, and most cases of SEF are caused by prolonged FS without neurological sequelae, where only the seizure is managed. However, some patients with SEF develop neurological emergencies, such as acute encephalopathy (AE; Hayakawa et al. 2016). In some cases, longer seizure duration can be a sign of AE, (Tang et al. 2024) and SEF can occur in the context of AE (Takanashi 2009). Therefore, SEF and AE are closely related. AE occurs in 400–700 children in Japan annually, and its incidence is reportedly high, especially in East Asia, suggesting a genetic background. However, infections (primarily viral, bacterial, or mycoplasmal) are the leading cause of AE in children (Mizuguchi et al. 2007, 2021). Most patients with SEF tend to have a favorable outcome without lasting sequelae. While most patients with SEF tend to have favorable outcomes without long‐term sequelae, the prognosis significantly differs when seizures occur in the context of AE, compared to those associated with FS. Many healthcare professionals may ponder whether such cases have any genetic predisposition or if there exists a shared underlying genetic background, involving a monogenic etiology common to developmental and epileptic encephalopathies (DEEs; Figure 1). In 2017, the International League Against Epilepsy introduced the concept of DEE (Scheffer et al. 2017). DEE is characterized by encephalopathy with intellectual disabilities associated with developmental delay or epilepsy, which is often resistant to antiseizure medicine. Genetic variants are often found in patients with DEE, owing to recent advances in genetic testing (Bartolini 2021).
With regard to the patients’ monogenic backgrounds of SEF and DEE, a Venn diagram is employed to illustrate the overlap of single gene variants. The left diagram represents scenarios where there is minimal sharing of background genetic variants between SEF and DEE. Conversely, the right diagram illustrates situations where the single gene variants in SEF and DEE are nearly identical.
To the best of our knowledge, no study has identified the different variants of background genes in patients with SEF. This may be attributed to the fact that many patients progress without sequelae, leading parents to decline genetic testing, or because FSs are relatively rare outside regions, such as East Asia, resulting in an insufficient number of cases to justify such testing.
First, in this study, we aimed to investigate disease‐associated gene variants in patients who have not yet been diagnosed with epilepsy and who developed SEF from fever‐induced seizures, as well as in those with DEE without a history of SEF. Second, we sought to compare the genetic variants between these two groups to elucidate the differences in the influence of each genetic variant on disease etiology.
Materials and Methods
2
Study Design and Patients
2.1
This retrospective clinical observational study was approved by the Ethics Committee of Kobe University (2022‐86). All the experiments were performed in accordance with the relevant guidelines and regulations of these institutions and the Code of Ethics of the World Medical Association (Declaration of Helsinki). Written informed consent was obtained from the parents of all patients. As of January 1, 2021, our department initiated genetic testing for patients with a medical history of SEF and DEE. The selection of genetic tests was determined by the attending physician, considering the patient's medical history, family history, and clinical findings. In this study, we retrospectively reviewed the medical charts of patients with a medical history of SEF and those with DEE who visited Kobe University Hospital and Kobe University‐affiliated hospitals (Hyogo Prefectural Kobe Children's Hospital and Takatsuki General Hospital) between January 1, 2021, and December 31, 2022, and requested a genetic diagnosis of SEF and DEE. We collected data on demographics, clinical presentation, treatments, imaging findings, and laboratory tests of these patients.
Definitions
2.2
FS was defined as seizures accompanied by fever (temperature ≥ 100.4°F or 38°C), without central nervous system infection, in infants and children aged 6–60 months, according to the American Academy of Pediatrics (Subcommittee on Febrile Seizures, American Academy of Pediatrics 2011). Seizure duration was defined as the time from seizure onset (according to information provided by caregivers) to the cessation of seizures (as confirmed by information provided by caregivers or emergency department physicians; Yamaguchi et al. 2018, 2020). SEF was defined as a seizure lasting for ≥ 30 min or a recurrent seizure lasting for ≥ 30 min without full recovery of consciousness triggered by fever. Although SE has previously been defined as a seizure or a series of seizures lasting ≥ 30 min (Berg et al. 2004), seizures lasting for 5 min have recently been classified as an operational definition of SE because such seizures often do not cease spontaneously (Brophy et al. 2012). In our study, a seizure duration of ≥ 30 min was adopted as SE because of the increased risk of mortality (Cheng 2016). “Febrile” was defined as a temperature of > 38°C (Yamaguchi et al. 2018). The diagnosis of AE was based on the Japanese Clinical Consensus and Guidelines for Acute Encephalopathy in Children (Committee of Guidelines for Acute Encephalopathy in Children 2016). In brief, AE was diagnosed when a patient had disturbed consciousness with an acute onset lasting at least 24 h (Committee of Guidelines for Acute Encephalopathy in Children 2016). Patients with obvious central nervous system infections, such as meningitis or encephalitis (defined by a cerebrospinal fluid cell count > 8 cells/µL), were excluded from the study (Committee of Guidelines for Acute Encephalopathy in Children 2016; Oikawa et al. 2024). In this study, SEF cases were defined as those presenting with status epilepticus induced by fever (suspected infection) and not yet diagnosed with DEE. In contrast, patients with DEE had never experienced status epilepticus during febrile episodes.
The term “DEE” includes developmental impairment related to both the underlying etiology, independent of epileptiform activity, and the epileptic encephalopathy (Riney et al. 2022). In this study, DEE was also defined as cases with no history of SEF. The diagnosis of epilepsy was based on the 2014 definition by the International League Against Epilepsy (Fisher et al. 2014), which includes the following criteria: (1) the occurrence of two or more unprovoked seizures more than 24 h apart; (2) the occurrence of one unprovoked seizure with a recurrence risk over the next 10 years comparable to the general risk after two unprovoked seizures; or (3) a diagnosis of an epilepsy syndrome.
Neurological performance at baseline and prognosis after onset were assessed using the Pediatric Cerebral Performance Category scale. On this scale, scores of 1, 2, 3, 4, 5, and 6 indicate normal function, mild impairment, moderate impairment, severe impairment, prolonged vegetative state, and death, respectively (Fiser 1992).
Commercial‐Based Genetic Testing
2.3
Patients 5, 7, and 30 were clinically suspected of having Dravet syndrome, and commercial gene panel testing was performed at the Kazusa DNA Research Institute (https://www.kazusa.or.jp/genetest/). This gene panel included SCN1A, SCN1B, SCN2A, and GABRG2.
Direct Sequencing and Subcloning
2.4
Patient 28 was clinically suspected of having PIK3CA‐related overgrowth syndrome, and direct sequencing was performed. Genomic DNA was extracted from the peripheral blood of the parents using the QuickGene‐Auto S DNA Blood Kit (Kurabo Industrial Ltd., Osaka, Osaka, Japan). Polymerase chain reaction (PCR) was performed using Tks Gflex DNA Polymerase (Takara Bio Inc., Kusatsu, Shiga, Japan), and direct sequencing was performed using the BigDye Terminator v1.1 Cycle Sequencing Kit and Applied Biosystems SeqStudio Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA). The primers used for PCR and direct sequencing are listed in Table S1. A mosaic variant was suspected in PIK3CA based on a sequence electropherogram, and subcloning analysis was performed. PCR products obtained using the HotStarTaq Plus Master Mix Kit (Takara Bio Inc.) were ligated into the pT7Blue T‐Vector (Novagen, Madison, WI) using a DNA Ligation Kit Ver. 2.1 (Takara Bio Inc.), and subcloning was performed using Escherichia coli HST08 Premium Competent Cells (Takara Bio Inc.). Colonies were selected using ampicillin and X‐gal, followed by colony PCR performed using the EmeraldAmp PCR Master Mix (Takara Bio Inc.) and primers (F: 5’‐TAATACGACTCACTATAGGG‐3’, R: 5’‐GTTTTCCCAGTCACGACGTTG‐3’). PCR products were analyzed by electrophoresis on a 1.5% agarose gel and direct sequencing with a BigDye Terminator v1.1 Cycle Sequencing Kit and Applied Biosystems SeqStudio Genetic Analyzer (Thermo Fisher Scientific).
Original Panel Sequencing
2.5
Genomic DNA was extracted from the peripheral blood of the parents using the QuickGene‐Auto S DNA Blood Kit (Kurabo Industrial Ltd.). The library was prepared with SureSelect PostPool Custom Tier2 (0.5–2.9 Mb) (Agilent Technologies, Santa Clara, CA). The 331 genes included in the panel have been previously reported (Oikawa et al. 2024) and are listed in Table S2. Sequencing was performed using the MiSeq platform (Illumina, San Diego, CA), and analysis was performed using SureCall v4.2.1.10 software (Agilent Technologies). Direct sequencing was performed to confirm candidate variants, which were evaluated according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines (Richards et al. 2015). Segregation analysis was performed by direct sequencing using gDNA derived from parents’ blood to confirm the presence of variants revealed in the probands. Therefore, the test for fatherhood was not performed.
Whole Exome Sequencing (WES)
2.6
Genomic DNA was extracted from the peripheral blood of the parents using the QuickGene‐Auto S DNA Blood Kit (Kurabo Industrial Ltd.). Whole‐exome sequencing was performed using NovaSeq 6000 (Illumina) and SureSelect Human All Exon V6 (Agilent Technologies). Reads were mapped to human genome assembly hg38 with the BWA‐MEM algorithm of Burrow–Wheeler Aligner v0.1.17 (Li and Durbin 2009). To obtain BAM files, PCR duplicates were marked with Picard MarkDuplicate v2.18.29 (http://broadinstitute.github.io/picard), and the base quality score was recalibrated using the Genome Analysis Toolkit (GATK) v4.3.0.0 (McKenna et al. 2010). Variant calling was then performed using the GATK Haplotype caller, and VCF files were obtained. The VCF files were annotated using ANNOVAR v2019Oct24 (Wang, Li, and Hakonarson 2010). Direct sequencing was performed to confirm the candidate variants found by WES, and the variants were evaluated according to the ACMG/AMP guidelines (Richards et al. 2015). Segregation analysis was performed by direct sequencing using gDNA derived from parents’ blood to confirm the presence of variants revealed in the probands. Therefore, the test for fatherhood was not performed. In patient 24, a variant was detected at the splice site of DEPDC5. Total RNA was extracted from the patient's blood using RiboPure RNA Purification Kit, blood (Thermo Fisher Scientific) and cDNA was obtained with SuperScript III Reverse Transcriptase (Thermo Fisher Scientific). Direct sequencing was performed on the obtained cDNA using Tks Gflex DNA Polymerase, BigDye Terminator v1.1 Cycle Sequencing Kit, and Applied Biosystems SeqStudio Genetic Analyzer to confirm the effect on splicing.
Microarray
2.7
Comparative genomic hybridization and single‐nucleotide polymorphism arrays were performed using GenetiSure Dx Postnatal Assay (Agilent Technologies). A SureScan Dx Scanner (Agilent Technologies) was used for scanning, and CytoDx (Agilent Technologies) software was used for analysis.
Statistics
2.8
The results are expressed as numbers. Fisher's exact test was used as appropriate to analyze the data. All analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). Statistical significance was set at p < 0.05.
Results
3
Clinical Background of the Patients With SEF and DEE
3.1
The clinical backgrounds of patients with SEF and DEE are shown in Tables 1 and 2, respectively. Fifteen patients were diagnosed with SEF (Table 1). The sex ratio was 9:6 (male:female). The median age at onset was 18 months (interquartile range [IQR], 13–37 months). Out of 15 patients diagnosed with SEF, five were finally diagnosed with AE (AESD, n = 3; hemorrhagic shock and encephalopathy syndrome, n = 2). The etiology of infection was identified in five cases. Generalized tonic‐clonic seizures (nine patients [60.0%]) and focal‐to‐bilateral tonic‐clonic seizures (three patients [30.0%]) were common types of seizures. The median duration of seizures was 120 (IQR, 62–168) min. Twenty‐seven patients were diagnosed with DEE (Table 2). The sex ratio was 14:13 (male:female). The age at onset was < 1 year in more than half of the patients with DEE (63.0%). Most patients had developmental delays or intellectual disabilities.
Results of Genetic Tests for the Patients With SEF and DEE
3.2
Genetic tests performed for each individual patient are shown in Table S3. Genetic variants explaining the clinical presentation were detected in four of the 15 patients with SEF and in 17 of the 27 patients with DEE. In patients with SEF, the variants were all found in SCN1A (n = 4). In patients with DEE, the variants were found in STXBP1 (n = 2), ALPL (n = 1), CDKL5 (n = 1), CSNK2B/TUBB (n = 1), DEPDC5 (n = 1), GABRB3 (n = 1), GNAO1 (n = 1), KCNB1 (n = 1), KCNQ2 (n = 1), PACS1 (n = 1), PIK3CA (n = 1), PTEN (n = 1), SCN1A (n = 1), SCN8A (n = 1), SHANK3 (n = 1), and TRRAP (n = 1; Table 3).
Thirteen of the 15 patients with SEF and 24 of the 27 patients with DEE underwent original panel sequencing. There was no significant difference in the number of original panel sequencings performed in both groups (p = 1.00; Fisher's exact test; Table 4). Three of 15 patients with SEF and 12 of 27 patients with DEE underwent whole‐exome sequencing. There was no significant difference in the number of WES analyses performed in both groups (p = 0.18; Fisher's exact test; Table 4). Two of the 15 patients with SEF and six of the 27 patients with DEE underwent microarray analysis. There was no significant difference in the number of microarrays performed in both groups (p = 0.69; Fisher's exact test; Table 4). Two of the 15 patients with SEF and two of the 27 patients with DEE underwent other genetic testing, including direct sequencing for PIK3CA and commercial‐based panel sequencing for Dravet syndrome. There was no significant difference in the number of other genetic tests performed in both groups (p = 0.61, Fisher's exact test; Table 4).
For Patients 7 and 30, the parents’ DNAs were not obtained; therefore, we could not confirm that the variants were de novo in their occurrence. The variant (NM_001165963.3:c.4398C>A,p.F1466L) detected in Patient 7 was not registered in the Genome Aggregation Database (gnomAD). Multiple in silico analyses (e.g., Mutation Taster, Polyphen‐2, SIFT, and CADD) predicted that the variant was damaging. A variant, in which the same amino acid residue is changed to serine (p.F1466S), is reportedly pathogenic (Lindy et al. 2018). Patient 7 was clinically diagnosed with Dravet syndrome, and no variants were identified in other genes associated with the syndrome, such as SCN1B, SCN2A, and GABRG2. Therefore, although the variant is classified as a variant of uncertain significance according to the ACMG guidelines, it was considered to be the likely cause of the disease.
The variant NM_001165963.3.4072T>C (p.W1358R) detected in patient 30 was previously reported as a de novo occurrence in a patient with Dravet syndrome (Zuberi et al. 2011). Additionally, this variant is not registered in the gnomAD database and was predicted to be damaging by multiple in silico analyses. Patient 30 was also clinically diagnosed with Dravet syndrome. As this variant has been previously reported as pathogenic, it was determined to be the cause of the disease in this case.
Comparison of the Results of Genetic Tests for the Patients With SEF and DEE
3.3
Four of 15 patients with SEF/AE and 17 of 27 patients with DEE had a genetic variant that explained their clinical presentation. The detection rate of genetic variants was higher in patients with DEE (63.0%) than in those with SEF (26.7%), although there was no statistically significant difference (p = 0.05, Fisher's exact test).
In both the SEF and DEE groups, no statistically significant differences were found between patients who tested positive for genetic mutations (genetic test+) and those who tested negative (genetic test−) across various clinical factors. Imaging abnormalities, such as those detected by computed tomography or magnetic resonance imaging, were observed in 50.0% of cases in both SEF groups (p = 1.0) and 13.3% versus 44.4% in the DEE group (p = 0.15). Past medical history showed a difference of 100% versus 45.5% in SEF (p = 0.10) and 100% versus 100% in DEE (p = 1.0). Developmental delays were present in 50.0% versus 36.4% in SEF (p = 1.0) and 100% versus 100% in DEE (P = 1.0). Similarly, family history showed no significant difference, with 25.0% versus 45.5% in SEF (p = 0.60) and 11.8% versus 10.0% in DEE (p = 1.0). These analyses were conducted using Fisher's exact test.
Discussion
4
We investigated the disease‐associated gene variants in patients with SEF and DEE. In addition, we compared the variants among patients with SEF and DEE to reveal the differences between SEF and DEE in terms of the influence of each genetic variant on the cause of the disease. The incidence of SEF is particularly low outside of East Asia, making it difficult to accumulate cases, and attention is often focused on environmental factors, such as the cause of infectious diseases. To the best of our knowledge, this is the first study to elucidate the different single genetic variants of SEF and compare them to those of DEE. The present study suggests that SEF may differ from DEE in terms of the diversity of single‐gene variants and the amount of variants that can be attributed to a specific gene.
Numerous genetic etiologies of DEE have been reported to date (Specchio and Curatolo 2021). Single or multiple genetic mutations combined with environmental factors are found in approximately 30%–70% of epilepsy cases (Wang et al. 2021; Çapan et al. 2024; Vetri et al. 2024). Our study identified causative genetic variants in 63% of DEE cases, aligning with previous studies. In the control group, several novel variants were found to be associated with DEE (patients 7, 9, 16, 24, 25, 26, 27, 31, 35, and 40), most of which were confirmed as de novo occurrences and classified as “pathogenic” or “likely pathogenic” according to the ACMG/AMP guidelines (Richards et al. 2015). Some of these variants were clinically and genetically important (Patients 16 and 17).
The SEF is closely related to the AE. Although some studies have reported a single‐gene mutation variant of SE with epilepsy (Wang et al. 2021; Neubauer and Hahn 2014; Bhatnagar and Shorvon 2015), mutation variants of SEF are scarce because many studies have focused on the source of fever, such as pathogenic microorganisms (Han and Han 2023). Contrastingly, AE is often an infection‐triggered emergency syndrome. It is assumed that the immune response, metabolism, and neuronal excitation are important triggers of AE (Mizuguchi et al. 2023). Furthermore, environmental factors, such as pathogenic microorganisms (e.g., influenza virus, human herpesvirus‐6), are important first triggers; however, genetic factors have also been reported to be the underlying etiology in some patients with AE. Regarding infection‐triggered AE, there are previously published case reports, case series, or studies on types of single‐gene variants within a particular disease (Saitoh et al. 2012, 2015; Shibata et al. 2020; Van et al. 2021; Kobayashi et al. 2010, 2012, 2013; Fukasawa et al. 2015; Neilson et al. 2009; Xavier et al. 2020; Denier et al. 2014; Ohashi et al. 2021; Mancardi et al. 2021; Shimada et al. 2018; Oláhová et al. 2018; Torisu et al. 2006; Isobe et al. 2022; Saito et al. 2022; Kurahashi et al. 2018; Tamhankar et al. 2020; Okuzono et al. 2019; Pezzani et al. 2018; Belal et al. 2018; Schmelzer et al. 2018; Merwick et al. 2013; Zhang et al. 2022; Schon et al. 2003; Ruitenbeek et al. 1995; Hidaka et al. 2006; Kara et al. 2022; Hanafusa et al. 2023). The genes encoding ion channels, that is, SCN1A and SCN2A, are the most commonly reported (Saitoh et al. 2012, 2015; Shibata et al. 2020; Van et al. 2021; Kobayashi et al. 2010; Fukasawa et al. 2015; Kobayashi et al. 2012; Hanafusa et al. 2023). In the present study, although many rare variants were found in DEE, only variants of SCN1A were found in SEF. Our study demonstrated that sodium channels are a critical factor in SEF. Interestingly, none of the patients with DEE in our study had a history of SEF, and the only gene common to both SEF and DEE cases was SCN1A. Therefore, we hypothesized that SEF and DEE would have different onset mechanisms. In DEE, there is a relatively strong genetic predisposition, while in SEF, environmental factors, such as the pathogenicity of infectious diseases, may play a role alongside the genetic predisposition associated with SCN1A mutations (Mizuguchi et al. 2007, 2021). Recent studies have reported associations between genetic polymorphisms and FSs or AE (Shibata et al. 2022; Skotte et al. 2022; Dimitrijevic et al. 2022). It is possible that the role of genetic polymorphisms in SEF will become clearer with further research. However, determining whether environmental factors or genetic predisposition exert a more significant influence requires additional investigation.
This study had some limitations. First, it had a retrospective, single‐center design, limiting the generalizability of its results. Second, as the convulsion time was estimated based on interviews with family members, the accuracy of reporting cannot be confirmed. Third, our study included a small number of patients because of its single‐center design. Despite these limitations, our study provides new insights into the pathophysiology of SEF and DEE.
In conclusion, analysis of patients with DEE revealed a wide variety of causative genes for DEE, whereas in SEF cases, only SCN1A variants were detected. Our study is the first to suggest that there is a difference in the detection rate of single gene variants of SEF, compared to that of the single gene variants of DEE, thereby implying that SEF may have a different pathophysiology in terms of single gene variants. Our findings can be further validated by accumulating additional databases of SEF cases to demonstrate the respective proportions of single gene variants amongst these diseases. Panel sequencing is highly useful for identifying genetic variants, but in some cases, pathogenic variants have been detected through WES rather than panel sequencing. As the cost of WES continues to decrease, it may become a more accessible and effective tool for genetic testing, offering broader diagnostic potential.
Author Contributions
Hiroaki Hanafusa: conceptualization, writing–original draft, methodology, data curation, investigation, formal analysis. Hiroshi Yamaguchi: conceptualization, investigation, funding acquisition, writing–original draft, methodology, validation, visualization, writing–review and editing, formal analysis, project administration, data curation, supervision. Naoya Morisada: data curation, writing–review and editing, methodology, resources, formal analysis. Ming Juan Ye: methodology, writing–review and editing, data curation, resources, formal analysis. Shizuka Oikawa: writing–review and editing, supervision, data curation. Shoichi Tokumoto: Writing–review and editing, supervision, data curation. Masahiro Nishiyama: writing–review and editing, supervision, data curation. Kandai Nozu: Writing–review and editing, supervision, data curation. Hiroaki Nagase: conceptualization, investigation, writing–review and editing, supervision, methodology, project administration, resources.
Ethics Statement
This retrospective clinical observational study was approved by the Ethics Committee of Kobe University (2022‐86). Informed consent was obtained from the parents of all patients. All the experiments in this study were conducted in accordance with the relevant guidelines and regulations, or in accordance with the Declaration of Helsinki. Written informed consent for publication of identifying images or other personal or clinical details was obtained from the parents or legal guardians of any participant under the age of 18.
Conflicts of Interest
The authors declare no conflicts of interest.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/brb3.70279
Supporting information
TABLE S1 Primer list for PIK3CA.
TABLE S2 The 331 genes included in the original gene panel.
TABLE S3 Genetic tests performed for each individual patient.
FIGURE S1 cDNA analysis of DEPDC5 in patient 24. We performed a cDNA analysis and found a short band in addition to the usual band in the patient, and exon 2 skipping was confirmed by sequencing analysis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bartolini, E 2021. “Inherited Developmental and Epileptic Encephalopathies.” Neurology International 13, no. 4: 555–568. 10.3390/neurolint 13040055.34842787 PMC 8628919 · doi ↗ · pubmed ↗
- 2Belal, H. , M. Nakashima , H. Matsumoto , et al. 2018. “De Novo Variants in RHOBTB 2, an Atypical Rho GT Pase Gene, Cause Epileptic Encephalopathy.” Human Mutation 39, no. 8: 1070–1075. 10.1002/humu.23550.29768694 · doi ↗ · pubmed ↗
- 3Berg, A. T. , S. Shinnar , F. M. Testa , et al. 2004. “Status Epilepticus After the Initial Diagnosis of Epilepsy in Children.” Neurology 63, no. 6: 1027–1034. 10.1212/01.WNL.0000138425.54223.DC.15452294 · doi ↗ · pubmed ↗
- 4Bhatnagar, M. , and S. Shorvon . 2015. “Genetic Mutations Associated With Status Epilepticus.” Epilepsy & Behavior 49: 104–110. 10.1016/j.yebeh.2015.04.013.25982265 · doi ↗ · pubmed ↗
- 5Brophy, G. M. , R. Bell , J. Claassen , et al. 2012. “Guidelines for the Evaluation and Management of Status Epilepticus.” Neurocritical Care 17, no. 1: 3–23. 10.1007/s 12028-012-9695-z.22528274 · doi ↗ · pubmed ↗
- 6Çapan, Ö. Y. , Z. Yapıcı , M. Özbil , and H. S. Çağlayan . 2024. “Exome Data of Developmental and Epileptic Encephalopathy Patients Reveals De Novo and Inherited Pathologic Variants in Epilepsy‐associated Genes.” Seizure: The Journal of the British Epilepsy Association 116: 51–64. 10.1016/j.seizure.2023.06.009.37353388 · doi ↗ · pubmed ↗
- 7Cheng, J. Y 2016. “Latency to Treatment of Status Epilepticus Is Associated With Mortality and Functional Status.” Journal of the Neurological Sciences 370: 290–295. 10.1016/j.jns.2016.10.004.27772779 · doi ↗ · pubmed ↗
- 8Denier, C. , L. Balu , B. Husson , et al. 2014. “Familial Acute Necrotizing Encephalopathy due to Mutation in the RANBP 2 Gene.” Journal of the Neurological Sciences 345, no. 1‐2: 236–238. 10.1016/j.jns.2014.07.025.25128471 · doi ↗ · pubmed ↗