Characteristics and Comparative Analysis of Six Mitogenomes of Genus Kiefferulus Goetghebuer, 1922 (Diptera: Chironomidae)
Dan Zhang, Wei-Dong Jin, Hai-Feng Xu, Xue-Bo Li, Yong-Wei Jiang, Dai-Qing Li, Xiao-Long Lin

TL;DR
This paper analyzes the mitochondrial genomes of six species of Kiefferulus, a genus of non-biting midges, to better understand their evolutionary relationships and genomic characteristics.
Contribution
The study provides the first comparative analysis of mitogenomes for six Kiefferulus species, offering insights into their phylogeny and mitochondrial genome evolution.
Findings
The six Kiefferulus mitogenomes contain 22 tRNAs, 13 PCGs, 2 rRNAs, and 1 CR with high AT content in the CR region.
Phylogenetic analysis using Maximum likelihood and Bayesian inference grouped the six species into a monophyletic clade.
ATP8 showed the highest evolutionary rate, while COX1 had the lowest among the protein-coding genes.
Abstract
Kiefferulus Goetghebuer, 1922 is an important genus within Chironomidae, known for its roles in aquatic ecosystems. The studies of this genus have mainly focused on morphological aspects, and the molecular data, especially high-throughput sequencing, are seriously inadequate, limiting our understanding of its evolution history. Herein, we sequenced the mitogenomes of six Kiefferulus species and analyzed the characteristics of the mitochondrial genome among these species. Furthermore, we reconstructed the phylogenetic relationship of the genus Kiefferulus based on two different methods: Maximum likelihood and Bayesian inference. In addition, the phylogenetic analysis of Kiefferulus exhibited that six newly obtained species were obviously distinguished from others. Our study provides a critical foundation for future research on the evolutionary biology of Chironomidae. Chironomidae is a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
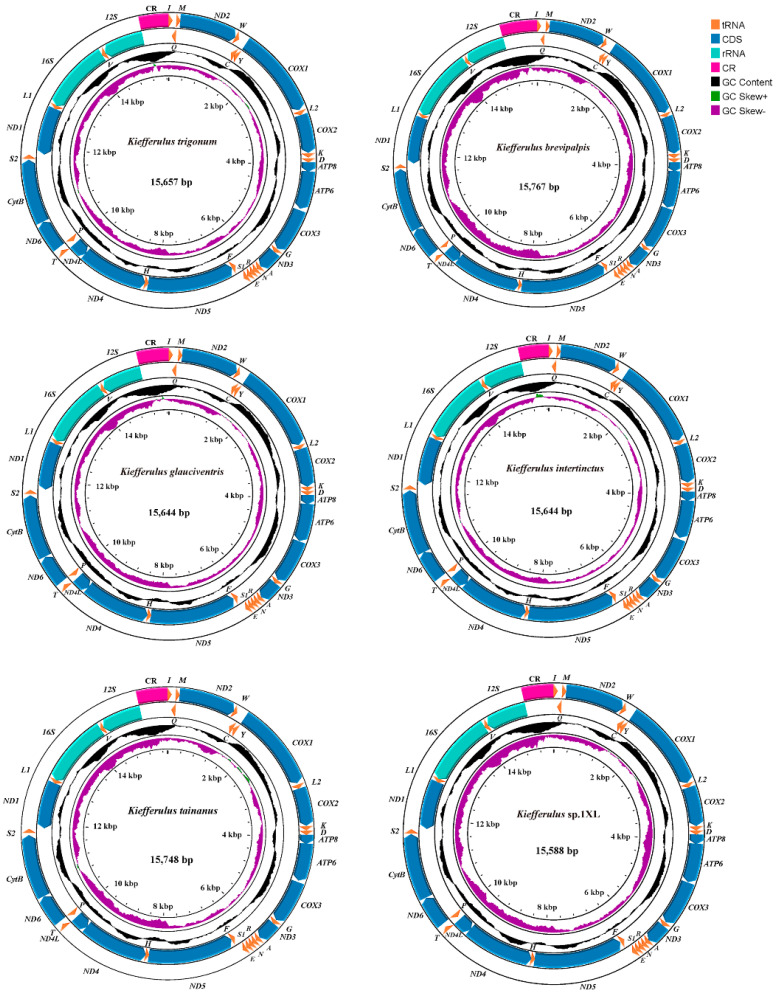 Figure 1
Figure 1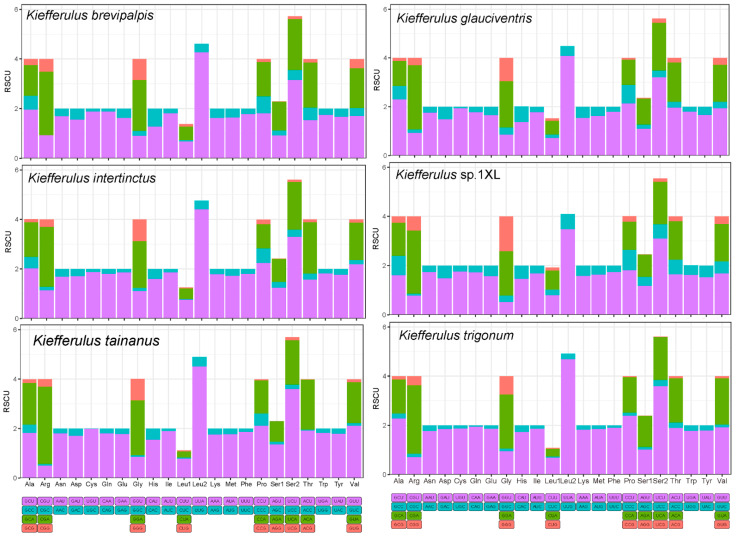 Figure 2
Figure 2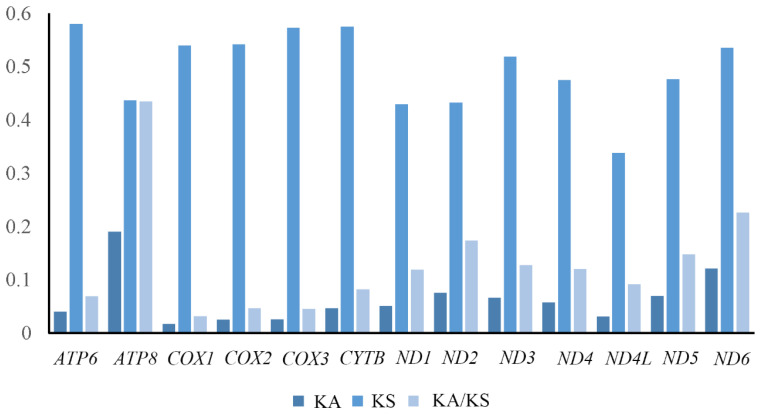 Figure 3
Figure 3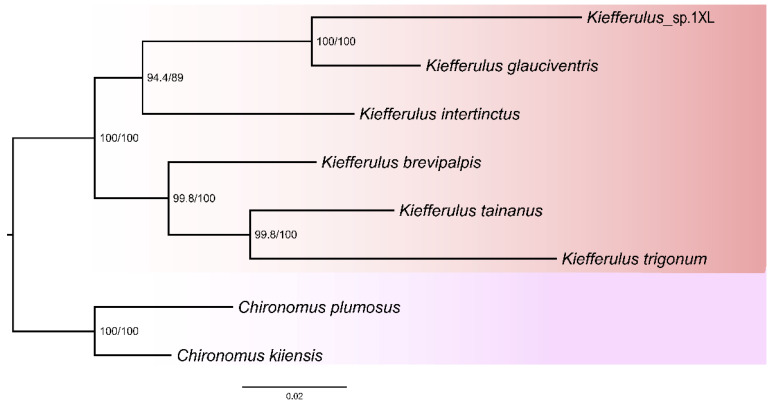 Figure 4
Figure 4- —National Natural Science Foundation of China
- —Shandong Provincial Natural Science Foundation
- —Sichuan Medical Law Research Center
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEnvironmental DNA in Biodiversity Studies · Protist diversity and phylogeny · Genomics and Phylogenetic Studies
1. Introduction
Chironomidae, as a cosmopolitan and important aquatic insect family, have a high diversity with more than 7500 described species worldwide [1]. Some Chironomids exhibit considerable resilience and resistance to environmental stressors [1,2,3,4]. The larvae of Chironomidae inhabit a wide range of environments, including freshwater, terrestrial, and semi-aquatic habitats [4]. Many species are highly sensitive to changes in environmental conditions, such as trophic state temperature, salinity, or acidity, making them useful indicator organisms for aquatic ecosystems [5].
Kiefferulus Goetghebuer, 1922 is a genus within the tribe Chironomini of the family Chironomidae, and can be distinguished from other genera by its characteristic hypopygium of males [6]. The larvae of this genus live in sediments of small- to medium-sized waterbodies, demonstrating a high level of adaptability to various environmental conditions; species of this genus can be used to assess the environment quality of tropical aquatic ecosystems [7,8]. Despite their ecological significance, research on this group is still inadequate, particularly in terms of morphological descriptions and molecular phylogenetic studies.
Mitochondrial genomes serve as significant molecular markers and have typically been utilized in investigations about phylogeny, evolutionary history, speciation, and phylogeography within insect taxa [2,9,10]. The maternal mode of inheritance, along with a high mutation rate and the ease of obtaining mitochondrial genomes, makes them particularly advantageous for the above research [3,11]. The insect mitochondrial genome usually ranges from 14,000 to 20,000 bp in length [9], including 2 ribosomal RNAs (rRNA), 22 transfer RNAs (tRNAs), 13 protein-coding genes (PCGs), and 1 non-coding control region (CR) [12]. Furthermore, the structural characteristics of mitogenomes can offer additional insights and corroborating evidence for taxonomic classification [2,3], and the number of complete mitochondrial genomes of insects has significantly increased. This advancement, driven by the rapid development of next-generation sequencing technologies, aids in resolving structural comparisons and understanding the evolutionary history of different groups [12,13,14].
In recent times, an increasing number of studies on the mitochondrial genomes of Chironomidae have greatly advanced research in systematics and evolutionary history. Additionally, the growing availability of mitogenomes of Chironomidae offers valuable opportunities to investigate the structure and evolutionary patterns of their mitochondria [2,3,14,15]. For example, Lin and his associates utilized mitochondrial genomes to examine the taxonomic characteristics and phylogenetic relationships of Prodiamesinae within the Chironomidae [2]. Their findings revealed that Prodiamesinae is a group of Orthocladiinae, providing additional insights into the evolutionary history of this group [2]. However, resources for the mitogenomes of the genus Kiefferulus are limited. Currently, only one mitogenome is available for Kiefferulus [16], which restricts our understanding of the phylogenetic relationships and evolutionary history of this group.
In this study, we sequenced, assembled, and annotated six mitogenomes of species within the genus Kiefferulus and analyzed their mitogenomic characters including their main features, evolutionary rates, and substitutions. In addition, we used eight mitochondrial genomes to reconstruct the phylogenetic relationships of Kiefferulus species. The results not only expand our understanding of the mitogenomic features of Chironomidae and Kiefferulus but also provide new information for the definition of the genus Kiefferulus.
2. Materials and Methods
2.1. Taxon Sampling and Sequencing
In this study, six species of the genus Kiefferulus were selected for sequencing analysis (Table 1). Cranston et al. (1990) synonymized the genera Nilodorumi Kieffer and Carteronica Kieffer with the genus Kiefferulus [17].Fang et al. reported the first mitogenome of the genus Kiefferulus; however, they mistakenly used the species name Nilodorum tainanus [16]. Thus, we used our newly sequenced data of K. tainanus for our analyses. In addition, we selected two species of Chironomus (Chironomus kiiensis: NC_069311.1; Chironomus plumosus: NC_069032.1) [16,18], which were closely related to the genus Kiefferulus as outgroups, based on a prior study of Chironomidae [6].
Herein, newly obtained samples were collected from China, Namibia, and New Caledonia during 2013–2021 (see detailed information in Table 1). All samples were deposited in 95% ethanol at −20 °C firstly for morphological examination and DNA extraction. X.L.L. identified all specimens, and all vouchers were stored at the College of Fisheries and Life, Shanghai Ocean University, Shanghai, China.
Qiagen DNeasy Blood & Tissue Kit was used to extract the whole-genomic DNA based on the manufacturer’s protocol. Qubit^®^ 2.0 Flurometer (ThermoFisher, Waltham, MA, USA) was used to measure the DNA concentration with Qubit^®^ DNA Assay Kit, and all the whole-genome DNA was then sent to the sequencing company (Berry Genomics, Beijing, China).
Sequencing libraries were generated by the Truseq Nano DNA HT sample preparation Kit (Illumina, San Diego, CA, USA), and the Illumina NovaSeq 6000 platform was used to sequence all raw data, generating 150 bp paired-end reads with an insert size of 350 bp. Short reads, low-quality reads, and adapters were removed from the raw data by Trimmomatic v0.32 (Jülich, Germany) [19].
2.2. Assembly, Annotation, and Composition Analyses
To guarantee precision, we employed two techniques for de novo assembly: (1) NOVOPlasty v3.8.3 (Brussel, Belgium) [20] was used to assemble the mitogenome with a k-mer size of 23–39 bp and (2) IDBA-UD v1.1.3 (Boston, MA, USA) [21] was performed to assemble with a “–mink 40 –maxk 120” parameter. Geneious 2020.2.1 [22] was employed to analyze and compare the mitogenome sequences obtained through the aforementioned methods and subsequently merge them into a single sequence. Annotation and analysis for the secondary structure of tRNAs were performed by tRNAscan SE 2.0 [23] and MITOS WebServer. MEGA X [24] was used to analyze the base composition and usage of mitogenomes. SeqKit v0.16.0 (Chongqing, China) [25] was utilized to evaluate the bias of the nucleotide composition of each gene. The AT-skew and GC-skew were computed as follows: AT-skew = (A − T)/(A + T), and GC-skew = (G − C)/(G + C). MEGA X was performed to calculate the AT-skew, GC-skew, and the relative synonymous codon usage (RSCU) of the newly sequenced species.
The rates of nonsynonymous substitution (Ka) and synonymous substitution (Ks) for each PCG were calculated using DnaSP 6.0 [26]. The online server CGview (https://cgview.ca/, accessed on 22 June 2024) was utilized to create the mitogenome map, which illustrates sequence features.
2.3. Phylogenetic Relationship
To reconstruct the phylogenetic relationships of the genus Kiefferulus, we used 2 rRNA and 13 PCG genes of Kiefferulus mitogenomes. MAFFT v7.450 (Osaka, Japan) [27] was applied to align the nucleotide and protein sequences with the L-INS-I method. Trimal v1.4.1 (Barcelona, Spain) [28] was used to trim sequences under the “-automated1” strategy. Finally, we generated five matrices to reconstruct the phylogenetic relationship of Kiefferulus, and FASconCAT-G v1.04 (Santa Cruz, CA, USA) [29] was applied to concatenate matrices: the (1) cds_rrna matrix included all PCGs and two rRNA nucleotide reads; (2) cds_faa matrix contained all PCG amino acid reads; (3) cds12_fna matrix contained all PCG nucleotide reads except the third codon positions; (4) cds_fna matrix included all PCG nucleotide reads; and (5) cds12_rrna matrix contained PCG nucleotide reads which removed the third codon positions and two rRNA genes.
We used Bayesian inference (BI) and Maximum likelihood (ML) to infer the phylogenetic relationships of the genus Kiefferulus for all matrices. In all ML analyses, the best-fitting substitution models were selected via ModelFinder [30] within IQ-TREE 2 (Canberra, ACT, Australia) [31]. In addition, to minimize the impact of long-branch attraction, we used the posterior mean site frequency (PMSF) [32] model with the ‘-m − mtART + C60 + FO + R’ command to reconstruct the phylogenetic relationship of the genus Kiefferulus based on the matrix cds_faa. BI trees were implemented via PhyloBayes-MPI (Montréal, QC, Canada) [33] with the site-heterogeneous mixture model ‘-m CAT + GTR’. For the BI analysis, the Markov chain Monte Carlo (MCMC) analysis was performed twice with 10,000,000 generations, while maxdiff < 0.3 stopped the MCMC analysis. A consensus tree was created by combining the remaining trees after discarding 25% of the initial trees from each run as burn-in. Finally, we used iTOL, an online website, to beautify all trees (https://itol.embl.de/upload.cgi, accessed on 15 May 2024).
3. Results and Discussions
3.1. Mitogenomic Structure
The Illumina NovaSeq 6000 platform generated 3 Gb raw reads for each sample. A total of six newly sequenced mitogenomes of the genus Kiefferulus were obtained here, and all of them were complete. All newly sequenced mitochondrial genomes were submitted in GenBank as PP884095–PP884099 and PP972215 (Table 1). The complete mitogenome of newly obtained species ranged from 15,588 (Kiefferulus sp.1XL) to 15,767 (Kiefferulus brevipalpis) bp in length; the unstable size of the CR was the main reason for the variation in mitogenome length (Table 2). As with all published insect mitochondrial genomes, all newly obtained sequences contained 37 typical genes, including 13 PCGs, 22 tRNAs, and 2 rRNAs (Figure 1). In general, the mitogenomic structure and nucleotide composition of newly obtained sequences displayed the typical characteristics of the Chironomidae [15,34,35]. The mitogenome characters of the newly sequenced species are shown in Figure 1.
The nucleotide composition of the newly obtained species exhibited similarities (Table 2), demonstrating the characteristic AT-biased composition that is commonly observed in Chironomidae and other insects [15,17,36]. The AT content of the newly obtained sequences ranged from 75.12% (Kiefferulus sp.1XL) to 79.44% (Kiefferulus trigonum), and the GC content ranged from 20.56% (K. trigonum) to 24.88% (K. sp.1XL). The AT-skew of all newly obtained mitogenomes was positive, while GC-skew was negative. The AT-skew of the newly sequenced mitogenomes ranged from 0.019 (K. trigonum) to 0.040 (K. brevipalpis), and the GC-skew ranged from −18.62 (Kiefferulus tainanus) to −0.186 (K. trigonum) (detailed information shown in Table 2).
3.2. Protein-Coding Genes, Codon Usage, and Evolutionary Rates
No significant disparities were observed in the size of tRNA, PCGs, and rRNAs among each species. The length of the 13 PCGs ranged from 11,220 (K. tainanus) to 11,621 (K. trigonum) bp. The AT content ranged from 73.10% (K. sp.1XL) to 78.24% (K. trigonum), while the GC content ranged from 21.76% (K. trigonum) to 26.90% (K. sp.1XL). By comparing and integrating previous research on Chironomidae, we found that the AT content at the third site of PCGs was notably higher than at the first and second sites [15] (Table 2). The skew metrics of all PCGs showed that both the AT-skew and GC-skew were negative. The AT-skew ranged from −0.192 (K. tainanus) to −0.175 (K. brevipalpis), and K. tainanus exhibited the highest GC-skew (1.38), while Kiefferulus sp.1XL showed the lowest GC-skew (−0.084) (for detailed information, see Table 2).
The CR size of the newly reported species ranged from 518 (K. trigonum) to 674 bp (K. brevipalpis). All 13 PCGs of the newly assembled mitogenomes exhibited the start codon ATN, which was typical for insect mitogenomes [9]. However, we identified several different start codon patterns: the start codon of ATP6, COX2, COX3, CYTB, ND4, and ND4L in five species was ATG; ATP8 was ATT in two species;; ND2 and ND6 were ATT in five species; COX1 and ND1 were TTG in five species; ND5 was GTG in one species; and so on, and for detailed information, see Figure S1. Although TAG or TAA were the common stop codons for this group, there were still several special stop codon patterns: ND4 had T as the stop codon in four species, and ND3 had TAG as the stop codon in one species. More detailed information is shown in Figure S2.
The relative synonymous codon usage (RSCU) patterns for the six mitochondrial genomes were predominantly congruent, as illustrated in Figure 2. The figure shows the RSCU values for all synonymous codons corresponding to the 22 amino acids, utilizing the 62 available codons in the 13 PCGs of Kiefferulus species. For the newly obtained Kiefferulus species, we found that the UUA, UUU, and AUU were the preferred codons. At the same time, Leu2, IIe, and Phe are frequently utilized amino acids, exhibiting the nucleotide composition preference for A/T. The codons RSCU > 2 of all newly sequenced species were as follows: UUA > UCU > CGA (Figure 2, Tables S1–S6).
The ratio of nonsynonymous (Ka) to synonymous (Ks) substitutions can be employed to gauge the proportion of non-neutral alterations compared to neutral ones, offering insights into the selective pressures acting on a protein-coding gene (PCG) [37,38]. In this study, the Ka/Ks of all PCGs was significantly lower than 1, ranging from 0.031 to 0.435. The evolution rates of the 13 PCGs were as follows: ATP8 > ND6 > ND2 > ND5 > ND3 > ND4 > ND1 > ND4L > CYTB > ATP6 > COX2 > COX3 > COX1 (Figure 3). Each PCG exhibited different levels of purifying selection. ATP8, ND6, and ND2 showed higher ω values, indicating relatively relaxed purifying selection pressure. Meanwhile, COX1 and COX3 exhibited hardly purifying selection.
3.3. Phylogenetic Relationships
We used five matrices cds_faa (3726 sites), cds_fna (11,178 sites), cds_rrna (13,359 sites), cds12_fna (7452 sites), and cds12_rrna (9633 sites) to infer the phylogenetic relationship of the genus Kiefferulus based on ML and BI approaches. Two different methods based on five matrices yielded 11 trees (Figure 4 and Figures S3–S12). All trees exhibited highly consistent topology. In our phylogenetic results, six Kiefferulus were distinctively differentiated from others and grouped in a monophyletic clade. The phylogenetic results indicated that K. trigonum was close to K. tainanus, and K. glauciventris was close to K. sp.1XL (Figure 4 and Figures S3–S12).
The phylogenetic relationships of Kiefferulus and its close group are still controversial. Sæther [39] analyzed the phylogenetic relationships of the tribe Chironomini using the characters of females and found that the Chironomus is the sister group of Kiefferulus. Martin [40] reconstructed the phylogenetic relationship among genera closely related to Chironomus based on molecular data; the results well supported the broader Kiefferulus concept, with Einfeldia identified as the sister group of the genus Kiefferulus. Song et al. [6] used two ribosomal genes and four protein-coding gene segments to reconstruct the phylogenetic relationship of Kiefferulus. In addition, Tang et al. [41] also analyzed the relationship among Kiefferulus and its closely related genera, but their relationships have not been fully resolved. Unfortunately, due to limited sample availability, we have not yet resolved the phylogenetic relationships among Kiefferulus and its closely related groups. In future studies, researchers can include more sampling of genera, both adults and larvae, using more efficient data to explore the evolutionary history of this group, such as the low-coverage whole-genome genome, which shows great promise for resolving relations in difficult groups [42,43].
4. Conclusions
This study successfully obtained the complete mitogenomes of six Kiefferulus species: K. sp.1XL, K. brevipalpis, K. glauciventris, K. intertinctus, K. tainanus, and K. trigonum. The structural characteristics of all newly obtained sequences were similar to published Chironomidae species. The base composition calculated results showed that A and T were obviously biased for all newly obtained mitogenomes, which were also similar to other published Chironomidae species. The nonsynonymous-to-synonymous substitution ratio showed that ATP8 displayed the highest evolution rate, and COX1 had the lowest evolution rate. Thus, our study provided more available mitogenome data for the study of Chironomini and Chironomidae. Our phylogenetic results among the species of the genus Kiefferulus showed that all trees exhibited highly consistent topology, and six Kiefferulus were distinctively differentiated from others and grouped in a monophyletic clade.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lencioni V. Cranston P.S. Makarchenko E.J. Recent advances in the study of Chironomidae: An overview J. Limnol.2018771610.4081/jlimnol.2018.1865 · doi ↗
- 2Lin X.L. Zhao Y.M. Yan L.P. Liu W.B. Bu W.J. Wang X.H. Zheng C.G. Mitogenomes provide new insights into the evolutionary history of Prodiamesinae (Diptera: Chironomidae)Zool. Scr.20225111913210.1111/zsc.12516 · doi ↗
- 3Lin X.L. Liu Z. Yan L.P. Duan X. Bu W.J. Wang X.H. Zheng C.G. Mitogenomes provide new insights of evolutionary history of Boreheptagyiini and Diamesini (Diptera: Chironomidae: Diamesinae)Ecol. Evol.202212 e 895710.1002/ece 3.895735646319 PMC 9130564 · doi ↗ · pubmed ↗
- 4Armitage P.D. Pinder L. Cranston P. The Chironomidae: Biology and Ecology of Non-Biting Midges Springer Science & Business Media Berlin/Heidelberg, Germany 201210.2307/5810 · doi ↗
- 5Giłka W. Zakrzewska M. Lukashevich E.D. Vorontsov D.D. Soszyńska-Maj A. Skibińska K. Cranston P.S. Wanted, tracked down and identified: Mesozoic non-biting midges of the subfamily Chironominae (Chironomidae, Diptera)Zool. J. Linn. Soc-Lond.202119487489210.1093/zoolinnean/zlab 020 · doi ↗
- 6Song C. Wang X. Bu W. Qi X. Morphology lies: A case-in-point with a new non-biting midge species from Oriental China (Diptera, Chironomidae)Zookeys 20209096710.3897/zookeys.909.3934732089635 PMC 7015952 · doi ↗ · pubmed ↗
- 7Epler J. Ekrem T. Cranston P.J. Cranston P.S. Epler J. The larvae of Chironomidae of the holarctic region-keys and diagnoses Insect Syst. Evol.20136638755610.5324/cjcr.v 0i 26.1656 · doi ↗
- 8Cranston P.S. The Chironomidae larvae associated with the tsunami-impacted waterbodies of the coastal plain of southwestern Thailand Bull. Raffles Mus.20075523124410.5281/zenodo.5333181 · doi ↗