Expressions of the satellite repeat HSAT5 and transposable elements are implicated in disease progression and survival in glioma
Sıla Naz KÖSE, Tutku YARAŞ, Ahmet BURSALI, Yavuz OKTAY, Cihangir YANDIM, Gökhan KARAKÜLAH

TL;DR
This study explores how satellite repeats and transposable elements in the genome are linked to glioma progression and patient survival.
Contribution
The study is the first to show that repeat/transposable element expression correlates with glioma progression and survival.
Findings
16 transposable elements were associated with slower disease progression in low-grade glioma patients.
HSAT5 satellite repeat and 22 transposons were linked to shorter survival in high-grade glioma patients.
HSAT5 co-occurred with genes related to chromosome segregation and nuclear division, suggesting a role in glioma pathogenesis.
Abstract
The glioma genome encompasses a complex array of dysregulatory events, presenting a formidable challenge in managing this devastating disease. Despite the widespread distribution of repeat and transposable elements across the human genome, their involvement in glioma’s molecular pathology and patient survival remains largely unexplored. In this study, we aimed to characterize the links between the expressions of repeat/transposable elements with disease progression and survival in glioma patients. Hence, we analyzed the expression levels of satellite repeats and transposons along with genes in low-grade glioma (LGG) and high-grade glioma (HGG). Endogenous transposable elements LTR5 and HERV_a-int exhibited higher expression in HGG patients, along with immune response-related genes. Altogether, 16 transposable elements were associated with slower progression of disease in LGG patients.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
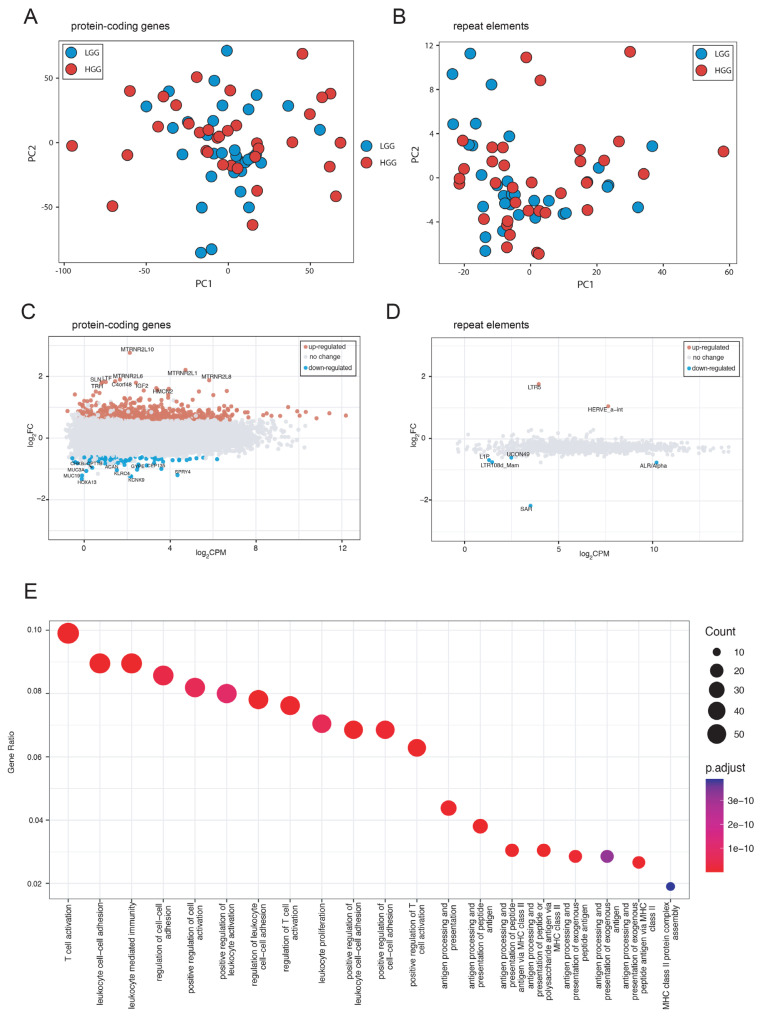 Figure 1
Figure 1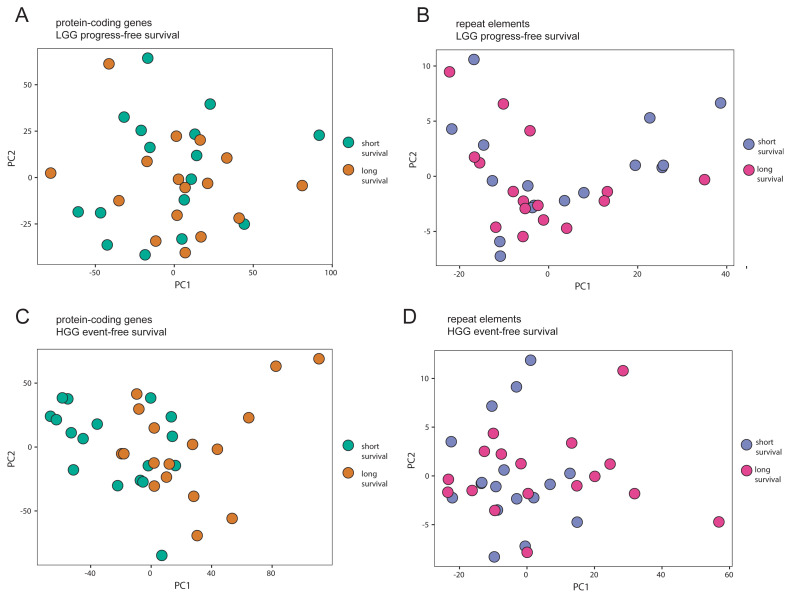 Figure 2
Figure 2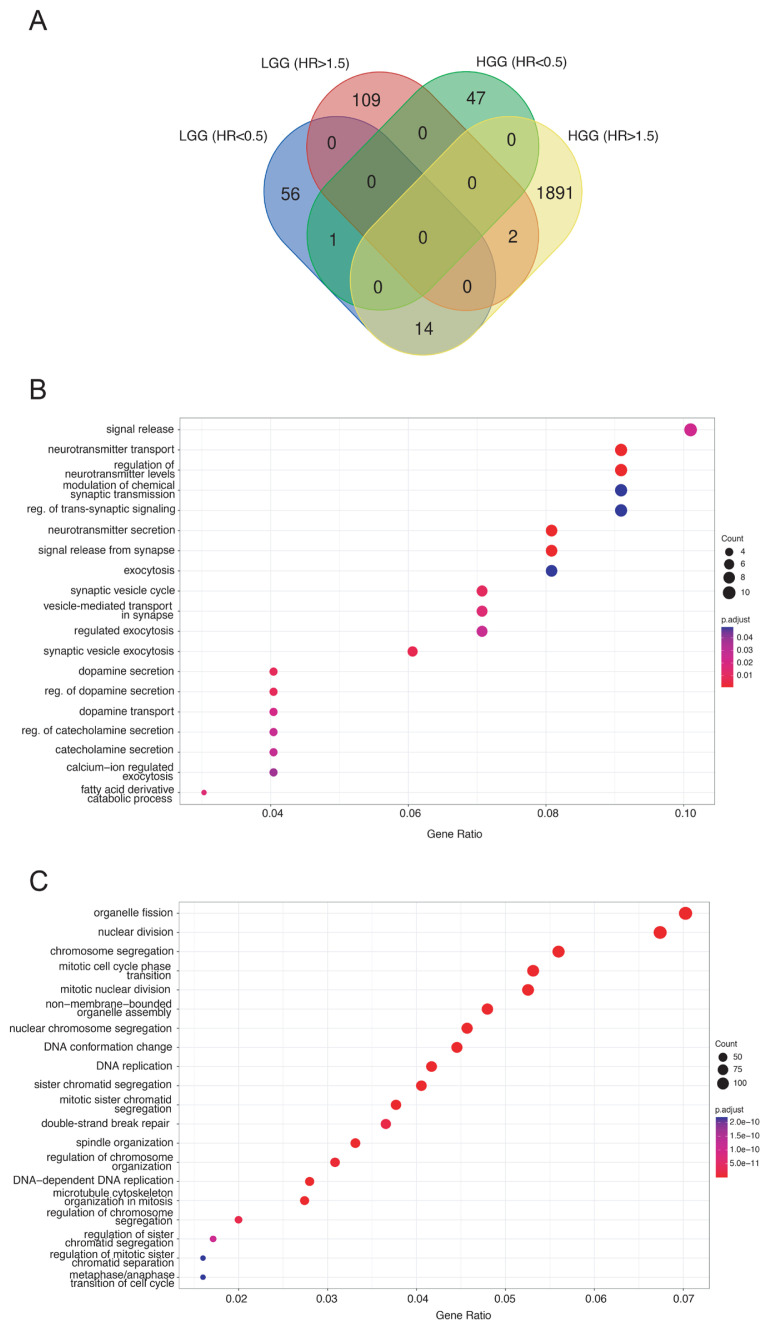 Figure 3
Figure 3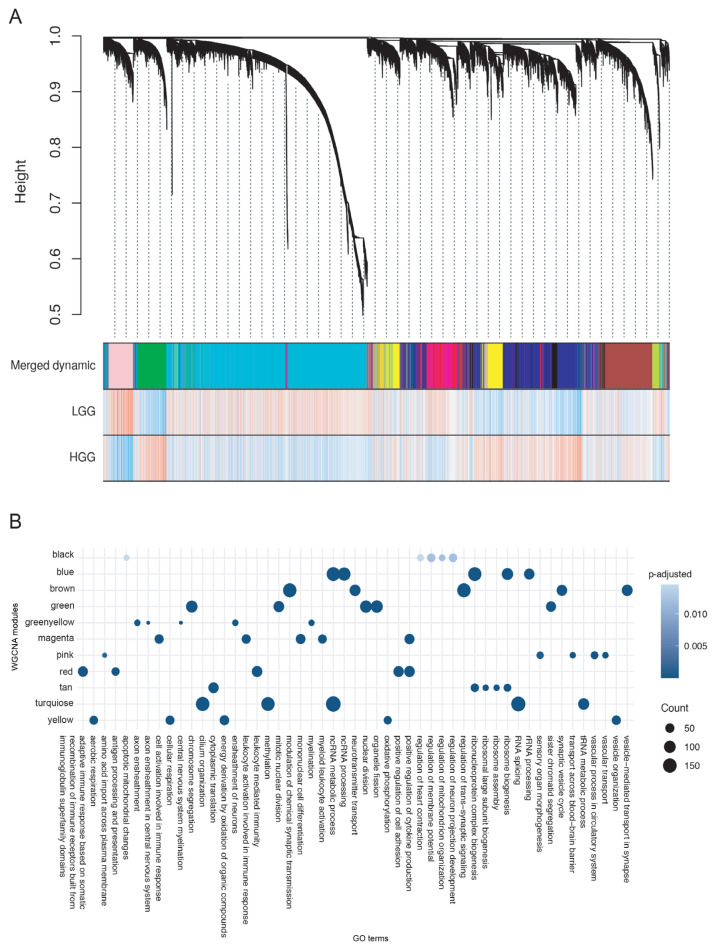 Figure 4
Figure 4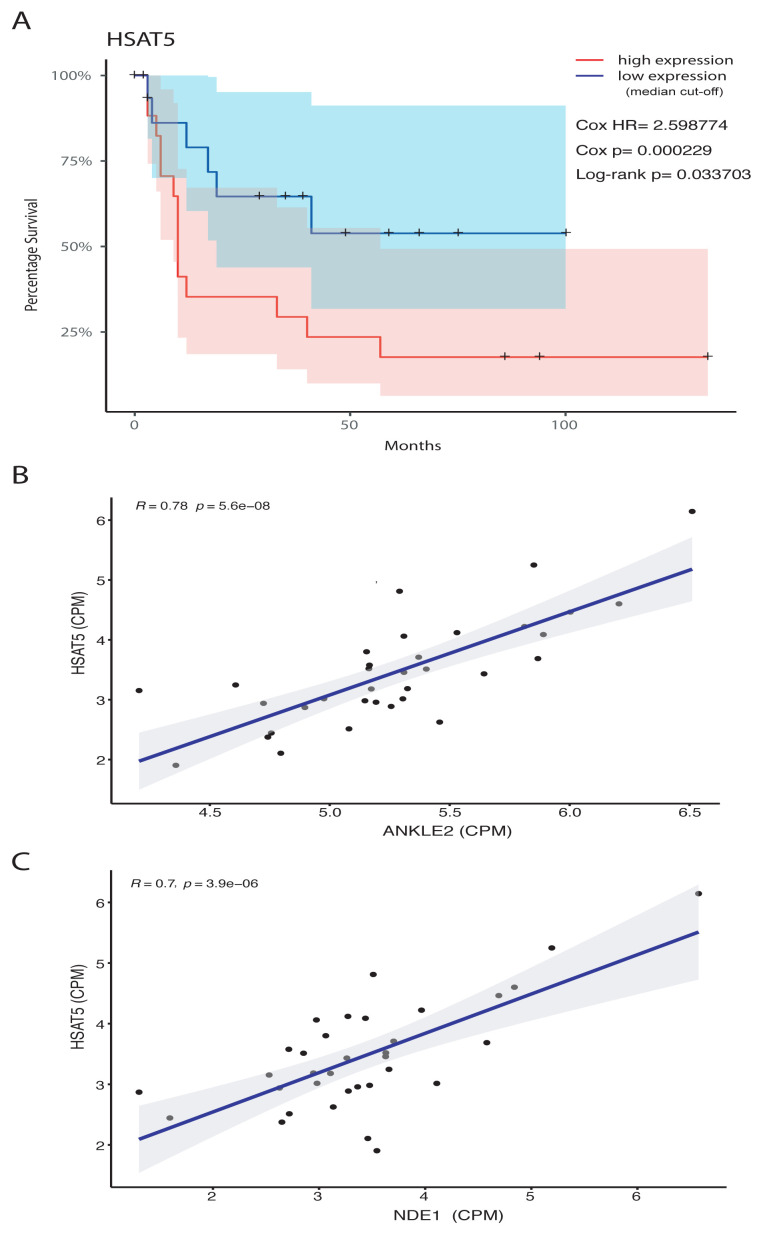 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA Research and Splicing · Ubiquitin and proteasome pathways · Microtubule and mitosis dynamics
1. Introduction
Gliomas are the most common type of central nervous system tumors representing approximately 80% of all malignant brain tumors (Ostrom et al., 2017). They can vary at genetic and epigenetic levels causing differences in tumor aggressiveness and hence a heterogeneous response to therapy (Noushmehr et al., 2010; Sturm et al., 2014; Sakthikumar et al., 2020; Garcia-Fabiani et al., 2021). Gliomas are histologically classified as astrocytic, oligodendroglial, or mixed (astrocytic and oligodendroglial) according to the cell type from which they originate (Louis et al., 2016). Importantly, they can be low-grade (LGG) or high-grade (HGG) according to their malignancy levels. LGGs typically arise de novo without any prior precursor or lower-grade lesions, and they have a better prognosis with approximately 75% five-year survival rates, while HGGs are grade III/IV gliomas generally differentiated from those of lower grades with a much lower five-year survival rate of only 5% (Stupp et al., 2009; Crespo et al., 2015; Teng et al., 2022). Even though gliomas could arise from different cell types as mentioned above, they exhibit similar molecular characteristics based on their grade (i.e. LGG or HGG) and mutational statuses (Arcella et al., 2020).
Numerous studies have attempted to unravel histological and molecular differences between LGG and HGG to facilitate the identification of potential biomarkers and therapeutic targets. LGGs are usually characterized by IDH, ATRX, and TP53 alterations, as well as CDKN2A hypomethylation and 1p/19q codeletion (Parsons et al., 2008; Brat et al., 2015; Louis et al., 2016), whilst progression to HGG can be characterized by PTEN mutations, EGFR amplifications and MGMT promoter hypermethylation (Wang et al., 2016; Brito et al., 2019). It is noteworthy that some of these alterations may have distinct and sometimes opposite prognostic implications in LGG and HGG. Several mutations were found to be associated with better outcomes in LGG whilst presenting worse outcomes in HGG (Eckel-Passow et al., 2015; Juratli et al., 2018). Moreover, dysregulations in signaling pathways, mutations in oncogenes and tumor suppressors, and impaired epigenetic mechanisms may lead to genomic instability and prompt the chromatin of a glioma cell for further malignant progression (Louis et al., 2016; Mackay et al., 2017). Pivotal changes in chromatin architecture were observed for various types of gliomas and they were related to tumor aggressiveness, as well as patient survival (Mack et al., 2019; Wang et al., 2021; Garrett et al., 2022; Yang et al., 2022). Further insights into the dynamics of these chromatin aberrations would therefore pave the way for designing more effective treatment strategies.
There are a multitude of layers that influence chromatin architecture, one of which certainly is the content of DNA. At least 50% of the human genome is made up of repeat elements (International Human Genome Sequencing Consortium, 2001) and their transcription levels reflect the status of chromatin (Probst et al., 2010; Jagannathan and Yamashita, 2017). Even though their exact contribution to subnuclear regulation is yet to be characterized, expression of repeats was generally linked to vital biological processes such as heat stress response (Porokhovnik et al., 2021) and cellular senescence (Karakülah and Yandım, 2020). More importantly, tightly regulated expressions of these elements were particularly implicated in embryonic development (Ulitsky et al., 2011; Yandım and Karakülah, 2019a). Whereas tandemly repeated arrays such as the satellites could affect kinetochore formation and hence chromosomal stability (Vos et al., 2006; Zhu et al., 2011), interspersed elements such as DNA transposons or retrotransposons (i.e. long interspersed nuclear elements [LINEs], short interspersed nuclear elements [SINEs], long terminal repeats [LTRs], and human endogenous retroviruses [HERVs]) may act as a mutagenic force (Lee et al., 2012; Helman et al., 2014). Therefore, not surprisingly, dysregulated transcription levels of both satellites and transposons were reported to be potentially disease-causing in various cancers including but not limited to breast cancer (Zhu et al., 2011; Yandım and Karakülah, 2019b), hepatocellular carcinoma (Karakülah and Yandım, 2021), and pancreatic cancer (Ting et al., 2011; Yandım and Karakülah, 2022).
Despite reported changes in global chromatin and genome regulation, there have only been limited investigations into the contribution of repeat elements to the molecular pathogenesis and prognosis of gliomas. Ohka et al. reported that hypomethylation events in LINE elements were associated with shorter overall survival in glioma patients (Ohka et al., 2011). In addition, it was indicated that transcriptions of Alu elements were posttranscriptionally edited in oligodendroglioma (Hwang et al., 2022). Another recent study focusing on endogenous retroviruses showed that 43 HERV elements were upregulated in HGG compared to healthy brain tissue with implicated effects on proximal gene regulation (Koso et al., 2012; Yuan et al., 2021). On the other hand, the specific types of repeats/transposable elements that could be associated with disease progression and survival in glioma have not yet been characterized. This could be partly because most of the published RNA sequencing (RNA-seq) data is prepared with a poly(A) bias in library preparation, which renders the analysis of repeat-arisen transcripts inconclusive (Solovyov et al., 2018).
To elucidate the particular links between repeat/transposable element expression and disease progression and survival in glioma patients, we employed a publicly available dataset (GSE184941) for which total RNA-seq had been performed without an mRNA bias on the tumor resection samples of 32 LGG and 34 HGG patients with IDH mutations (Supplementary file 1: Table S1) (Xiao et al., 2022; Khan et al., 2023). This was the only available relevant dataset that allowed the quantification of total transcripts arisen from repeat elements and transposons whilst also presenting patient survival attributes. We identified differentially expressed satellite repeats and transposons between LGG and HGG samples. We also performed survival analyses considering the expressions of all human RepBase repeats (Bao et al., 2015) with Cox-regression and log-rank tests (Bewick et al., 2004), revealing a subset of repeats that are potentially important for LGG to HGG progression and survival.
2. Results
2.1. Identification of differentially expressed genes and repeat elements in HGG in comparison to LGG
To uncover the differences in gene and repeat/transposon expression, we quantified total RNA transcripts and performed a principal component analysis (PCA) (Figure 1 and Supplementary file 2: Figure S1). Even though a general distinction was observed in PCA analysis both for protein-coding gene expression and repeat arisen transcription levels between LGG and HGG samples (Figures 1A and 1B), the distribution of read percentages across patients were slightly different (Supplementary file 2: Figure S1A) with small changes in the abundance of repeat types giving rise to transcripts (Supplementary file 2: Figure S1B and S1C). When we applied a threshold of | log_2_(FC)| >0.6 | and false discovery rate (FDR) < 0.05, we identified 605 differentially expressed genes (Figure 1C and Supplementary file 3: Table S2) and 7 repeats (Figure 1D and Supplementary file 3: Table S3) in HGG samples when compared to LGG samples. A gene ontology analysis revealed that differentially expressed genes were mostly associated with biological processes implicating immunological response and cell-cell adhesion (Figure 1E, Supplementary File 4: Table S4), aligning well with previous literature (Xu et al., 2019). Retrotransposons LTR5 and HERVE_a-int were upregulated in HGG samples suggesting a possible role for them in the progression from LGG to HGG.
2.2. Identification of gene and repeat expressions implicated in disease progression and survival
Gene expression signatures that influence survival times of patients provide invaluable information on the carcinogenesis and metastasis process in cancer (Sahu et al., 2021). Therefore, they have the potential to open new treatment avenues. To provide a detailed picture of the survival-related expression events where repeat-arisen transcripts were also considered in glioma, we performed a Cox regression analysis followed by a log-rank test (with a median progression/survival time cut-off) not only for the survival of HGG patients but also for the progression of LGG patients (to HGG). A PCA analysis showed a more pronounced separation between shorter and longer survival in the event-free survival analysis of HGG samples as opposed to progress-free survival analysis of LGG samples (Figure 2).
Based on progress-free survival analysis in LGG samples, we identified 71 genes and 16 repeats (Cox Hazard Ratio [HR] < 0.5, Cox p-value < 0.05 and log-rank p-value < 0.05) that were associated with slower LGG to HGG progression (Table 1, Supplementary file 5: Tables S5 and S7). On the other hand, 111 genes, but none of the repeats, (Cox HR > 1.5, Cox p-value < 0.05 and log-rank p-value < 0.05) were found to be related to faster LGG to HGG progression (Table 1, Supplementary file 5: Tables S6 and S8). In the samples where progression to HGG took place, event-free (event: death) survival analysis revealed 48 genes, but no repeats (Cox HR <0.5, Cox p-value<0.05 and Log-rank p-value<0.05) were associated with longer survival (Table 1, Supplementary file 6: Tables S9 and S11). Interestingly, 1907 genes and 23 repeats (Cox HR >1.5, Cox p-value<0.05 and Log-rank p-value<0.05) were linked to shorter event-free survival in HGG (Table 1, Supplementary file 6: Tables S10 and S12). Only one gene (CASS4) was associated with longer progress-free survival in LGG, as well as longer event-free survival in HGG (Figure 3A). On the other hand, two genes (GPR156 and AGAP1), but no repeats, were associated with shorter survival both in progress-free analysis of LGG samples and event-free analysis of HGG samples at the same time. Intriguingly, 14 genes (IPO9, PLK1, DCAF4L1, ZNF345, LRRC75A, RAD18, BMP8A, LRR1, SETD6, AK2, MAP3K12, LMBR1L, ERCC6L, ZNF776) and one transposon (LTR18C) were related with longer time for LGG-to-HGG progression, yet in opposition, they were linked to shorter event-free survival in HGG samples. Gene ontology analyses revealed that LGG-to-HGG progression could be attributed to neurotransmitter release (Figure 3B, Supplementary file 7: Table S13), whereas survival in HGG could be related to various cell cycle events including spindle organization, chromosome segregation, and nuclear division (Figure 3C, Supplementary file 8: Table S14).
2.3. Weighted gene co-expression network analysis (WGCNA) reveals the expression of HSAT5 satellite as a potential key player in HGG
In order to further elucidate specific gene expression correlation patterns that could be distinctly related to LGG or HGG and to provide a better insight into the cooperation of repeat expression in these patterns, we performed weighted gene co-expression network analysis (WGCNA) (Zhang and Horvath, 2005; Cantone and Fisher, 2013). Our pipeline contained a pool approach where not only the transcript levels of genes but also those of repeats were analyzed altogether. Overall, 13 modules that contained genes and repeats were identified and these were represented with different colors (Figure 4A, Supplementary file 9: Table S15). Tan module was specifically emphasized in LGG samples, and the green module was particularly associated with HGG. Whereas the tan module contained no repeats but included 185 genes, the green module comprised only the satellite repeat HSAT5, no transposons, and 981 genes. The majority (987) of repeat elements/transposons fell into turquoise module, which was emphasized in LGG samples and where all 16 repeats linked to longer LGG progress-free survival (Table 1) were also present. The blue module, on the other hand, was pronounced in HGG and contained 5 repeats, among which only L1M2a was linked to event-free survival in HGG. Greenyellow module was slightly pointing to LGG, and it contained only one repeat.
When a gene ontology analysis was run for all of the WGCNA modules, genes related to ribosome biogenesis and protein translation seemed to be enriched in the tan module (Figure 4B, Supplementary file 10). Interestingly, the green module displayed an enrichment in genes that were linked to nuclear division, chromosome segregation, and double-strand break repair. Turquoise module, where the majority of repeats reside, was enriched with genes linked to cilium organization, methylation and ncRNA metabolic process, splicing and tRNA metabolic process. The blue module was as well in an enrichment with ncRNA processing and ribosome biogenesis genes. On the other hand, the greenyellow module was related to myelination and neuron ensheathment.
Our interesting findings on HSAT5 satellite repeat which was linked to shorter event-free survival in HGG (Figure 5A) and the fact that it appeared in the same WGCNA module (green) with genes that were linked to similar biological phenomena (i.e. nuclear division and chromosome segregation) presented in Figure 3C for HGG event-free survival, prompted us to run a Pearson correlation analysis for the expression levels of HSAT5 and all protein-coding genes in the genome (Supplementary file 11). We found that 310 genes exhibited moderate-to-high level of correlation (r > 0.5) with HSAT5 expression. Strikingly, 195 (63%) of these 310 genes were among those significantly linked to shorter HGG event-free survival as presented in Table S10 (Supplementary file 6). A thorough literature search also showed that most of these genes were reported to be influential in glioma (Table 2). Some of these HSAT5-correlated genes including but not limited to ANKLE2 (Figure 5B) and NDE1 (Figure 5C) could be involved in the regulation of HSAT5 expression due to their crucial functions on chromatin (Chomiak et al., 2022; Sebastian et al., 2022).
3. Discussion
The complexity of molecular pathogenesis underlying gliomas makes them refractory to current treatment strategies. Notably, most of the patients that die due to gliomas suffer from HGG, which in most cases stem from a preceding LGG (Bogdańska et al., 2017). Therefore, understanding the particular events that cause progression to HGG from LGG is important in terms of projecting more insightful approaches toward gliomas. In this study, we uncovered not only differentially expressed protein-coding genes but also transcripts arisen from satellite repeats and transposons in HGG as opposed to LGG, shedding further light into disease progress. We found that genes related to immune response were differentially expressed in HGG as opposed to LGG, agreeing well with previous reports (Gabrusiewicz et al., 2016; Lu et al., 2019; Xiao et al., 2022; Khan et al., 2023). This could be a projected outcome due to immune cell infiltration as reported before (Domingues et al., 2016; Lin et al., 2021). Another interesting finding was the upregulated expression (in HGG) of HERV_a-int and LTR5 transposon, the latter of which was previously linked to cancer (Lee et al., 2012) and human preimplantation development (Yandım and Karakülah, 2019a).
Our progress-free survival analysis of LGG samples on gene expressions revealed that genes related to the regulation of neurotransmitter release, dopamine secretion and exocytosis were linked to faster progression to HGG. Even though genes within this context were not reported in some of the studies that analyzed clinical samples (Wang et al., 2020; Zhang et al., 2020), in vitro and animal studies previously suggested dopamine release as a potential factor in glioma (Li et al., 2014); however, its effect was not entirely understood (Lan et al., 2017). Another interesting finding in our study was that all repeat elements associated with LGG-to-HGG progression were those that had a hazard ratio of less than 0.5. In other words, transposon expression could potentially slow down the progression to HGG. This could be due to increased antigenicity and better immune response caused by transposon expression as suggested earlier (Kong et al., 2019; Yuan et al., 2021). In opposition, event-free survival analysis in HGG revealed that none of the repeat elements were associated with longer survival. However, it identified one satellite (i.e. HSAT5) and 23 transposons linked to shorter survival. Though this result suggests that repeat element expression may lead to opposite consequences in LGG and HGG, only one repeat (i.e. LTR18C) appeared in the survival analyses of both LGG (HR<0.5) and HGG (HR>1.5) groups. In line with this, the genes linked to shorter survival in HGG were also pointing out a different context as opposed to those in LGG. Genes with HR>1.5 were mostly linked to chromosome segregation and nuclear division, generally agreeing well with previous literature (Bredel et al., 2005; Phillips et al., 2006; Murat et al., 2008; Q. Wang et al., 2017).
In an effort to dissect the impact of repeat element expression on the molecular pathogenesis and survival in glioma, we not only presented LGG progress-free survival related repeats and HGG event-free survival linked repeats but also differentially expressed repeats in HGG samples in comparison to LGG. Yuan et al. (2021) reported differentially expressed repeats in tissue samples of HGG (GBM) patients as opposed to normal brain samples; however, the data they employed was prepared with an mRNA bias only detecting poly(A)+ transcripts. Our study utilizes sequencing data obtained from total RNA samples without the poly (A) bias (Khan et al., 2023) as previously recommended (Solovyov et al., 2018). Notably, LTR5 was reported to be upregulated both in a previous study (Yuan et al., 2021), which compares the HGG tissue with normal tissue, and in our study that compares with the HGG tissue with LGG, even though no survival related effect was detected. Another element we detected in HGG event-free survival analysis was HERVK3-int, which was reported to be upregulated in HGG samples with a potential effect on disease progression (Shah et al., 2022). Our holistic approach identified many other repeats/transposons previously unmentioned in the glioma literature. Perhaps, the most interesting one among these is the satellite repeat HSAT5, which predominantly resides in pericentromeric regions of chromosomes (Hillier et al., 2005; Hubley et al., 2016) and was also reported to be upregulated in cellular senescence (Karakülah and Yandım, 2020). Though no studies so far reported HSAT5 within the context of cancer, other satellites such as HSATI (Zhu et al., 2011), HSATII (Ting et al., 2011), and GSATII (Karakülah and Yandım, 2021) were linked to the molecular pathologies of solid cancers. Satellite repeats are known to play pivotal roles in maintaining heterochromatin architecture and kinetochore formation (Rošić et al., 2014; Nishibuchi and Déjardin, 2017) and their abnormal expressions were linked to genomic catastrophes in cell division (Zhu et al., 2011; Bersani et al., 2015; Kishikawa et al., 2016). Their usage as biomarkers is also being valuably considered (Özgür et al., 2021). Our results point out HSAT5 satellite repeat as a potential prognostic biomarker in HGG survival. More importantly, its cooperative expression levels with HGG-specific genes that are known to involve in chromosome segregation and nuclear division could implicate its potential in the molecular pathology of this devastating disease. The relationship of HSAT5 DNA or RNA with chromatin-related factors such as NDE1 and ANKLE2 could illuminate the mechanism of HSAT5 expression further.
4. Conclusion
In this study, we performed a comprehensive analysis on the expressions of repeat elements covering both satellite repeats and transposons to figure out those implicated in glioma progression and survival. Our results interestingly revealed that the expressions of transposable elements could be linked to slower LGG to HGG progression, yet this beneficial effect is overturned once the progress to HGG is manifested. One explanation for the beneficial effect in LGG could be the improved antigenicity caused by repeat elements, which provides better immune response. On the other hand, once the progression to HGG takes place, repeats could be contributing to dysregulated events in chromosome segregation and cell cycle as mostly the results on HSAT5 satellite repeat here suggested. Results presented here could serve as a guide for further experimental work in understanding the molecular basis of gliomas further.
5. Methods
5.1. Data acquisition and preprocessing
We obtained raw sequencing data from the Sequence Read Archive (SRA) database (Leinonen et al., 2011) for 66 glioblastoma patients (SRA accession: SRP339173; GEO accession: GSE184941) using the “fastq-dump -gzip -skip-technical -readids -dumpbase -clip -split-3” command of the SRA Tool Kit v.2.9.0. We collected the GRCh38 human genome assembly and comprehensive gene annotation (release 34) in gene transfer format from the GENCODE1 website (Frankish et al., 2021). We obtained genomic coordinates of repeat instances across the genome from the RepeatMasker2 website. We aligned paired-end reads from each tumor sample to the human reference genome using the Rsubread package v1.34.7 (Liao et al., 2014) in the R statistical computation environment with default parameters. We used SAMtools suite v1.3.1 (Li et al., 2009) to sort and index the binary alignment map files. We counted reads using the featureCounts function of the Rsubread package (Liao et al., 2014), which allowed us to differentiate between exonic and repetitive regions. We merged repeat and gene counts into a single expression matrix, and calculated counts per million (CPM) values for each gene and repeat element across all tumor samples. We excluded any gene or repeat element that was not expressed above 1 CPM in at least 25% of the tumor samples from further analysis. Besides, when filtering for HGG and LGG samples, IDH mutants and percentage of total mapped reads greater than 70 were included in both samples. To improve the accuracy of estimated repeat element expressions, we only considered sequencing reads that overlapped nonexonic regions and uniquely mapped to DNA, LINE, SINE, LTR, and satellite repeat regions.
5.2. Coexpression network analysis of protein-coding genes and repeat elements
To perform coexpression network analysis, we used the R package WGCNA v1.47 (Langfelder and Horvath, 2008). Adjacencies between all protein-coding genes and repeats across samples were calculated. In this step, CPM values of genomic features were used as input. We designated the soft threshold value as 12 and the average linkage hierarchical clustering method was selected for grouping the genomic features with similar expression patterns. Additionally, the dynamic tree cut algorithm (Langfelder et al., 2008) was utilized to determine network modules and minimum network module size was set to 30. Gene ontology (GO) enrichment analysis of each network module was performed with the R package clusterProfiler v3.18.0 (Yu et al., 2012).
5.3. Statistical analysis and graphical representation
Statistical analysis and graphical representation were performed using the R environment3 for the LGG and HGG samples. Differential expression (DE) analysis of genomic features was performed using the edgeR package (version 3.28.2) (Robinson et al., 2010). We obtained a PCA plot of samples to visualize clusters of LGG and HGG samples based on their similarities (quantile median = 0.50). We used the log rank test (logrank_test function of the coin package, version 1.4.1) (Royston et al., 2019) to test the statistical significance of survival time differences between groups and the Cox regression analysis (coxph function of the survival package, version 3.2.11) to estimate hazard ratios (HR). The Kaplan-Meier curves were drawn using the survfit function to show the probabilities of survival for a certain time interval. We used the ggplot2 package (version 3.3.6) to create other graphical representations.
Supplementary Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Apridita Sebastian W Shiraishi H Shimizu N Umeda R Lai S 2022 Ankle 2 deficiency-associated microcephaly and spermatogenesis defects in zebrafish are alleviated by heterozygous deletion of vrk 1 Biochemical and Biophysical Research Communications 624 95 101 10.1016/j.bbrc.2022.07.070 35940133 · doi ↗ · pubmed ↗
- 2Arcella A Limanaqi F Ferese R Biagioni F Oliva MA 2020 Dissecting molecular features of gliomas: genetic loci and validated biomarkers International Journal of Molecular Sciences 21 2 10.3390/ijms 21020685 PMC 701419031968687 · doi ↗ · pubmed ↗
- 3Baghbani F Raoofian R Hasanzadeh NM Hamzehloei T Soukhtanloo M 2013 Identification of novel hypoxia response genes in human glioma cell line a 172 Iranian Journal of Basic Medical Sciences, 16 5 675 82 23826488 PMC 3700041 · pubmed ↗
- 4Bao W Kojima KK Kohany O 2015 Repbase Update, a database of repetitive elements in eukaryotic genomes Mobile DNA 6 11 10.1186/s 13100-015-0041-9 26045719 PMC 4455052 · doi ↗ · pubmed ↗
- 5Balvers RK Kleijn A Kloezeman JJ French PJ Kremer A 2013 Serum-free culture success of glial tumors is related to specific molecular profiles and expression of extracellular matrix-associated gene modules Neuro-Oncology 15 12 1684 95 10.1093/neuonc/not 116 24046260 PMC 3829587 · doi ↗ · pubmed ↗
- 6Bersani F Lee E Kharchenko PV Xu AW Liu M 2015 Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer Proceedings of the National Academy of Sciences of the United States of America 112 49 15148 15153 10.1073/pnas.1518008112 PMC 467901626575630 · doi ↗ · pubmed ↗
- 7Bewick V Cheek L Ball J 2004 Statistics review 12: survival analysis Critical Care (London, England) 8 5 389 394 10.1186/cc 2955 15469602 PMC 1065034 · doi ↗ · pubmed ↗
- 8Bogdańska MU Bodnar M Piotrowska MJ Murek M Schucht P 2017 A mathematical model describes the malignant transformation of low grade gliomas: prognostic implications Plo S One 12 8 e 0179999 10.1371/journal.pone.0179999 28763450 PMC 5538650 · doi ↗ · pubmed ↗