The genome sequence of the Barred Chestnut moth, Diarsia dahlii (Hübner, 1813)
David C. Lees, Jose Martinez, Annabel Whibley

TL;DR
This paper provides the genome sequence of the Barred Chestnut moth, including its chromosomes and mitochondrial DNA, along with gene annotations.
Contribution
The study presents a high-quality genome assembly and gene annotation for Diarsia dahlii, including sex chromosomes and mitochondrial DNA.
Findings
The genome assembly spans 683.0 megabases and includes 32 chromosomal pseudomolecules.
Gene annotation identified 13,177 protein coding genes using Ensembl.
The mitochondrial genome is 15.36 kilobases in length.
Abstract
We present a genome assembly from an individual female Diarsia dahlii (the Barred Chestnut; Arthropoda; Insecta; Lepidoptera; Noctuidae). The genome sequence is 683.0 megabases in span. Most of the assembly is scaffolded into 32 chromosomal pseudomolecules, including the Z and W sex chromosomes. The mitochondrial genome has also been assembled and is 15.36 kilobases in length. Gene annotation of this assembly on Ensembl identified 13,177 protein coding genes.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1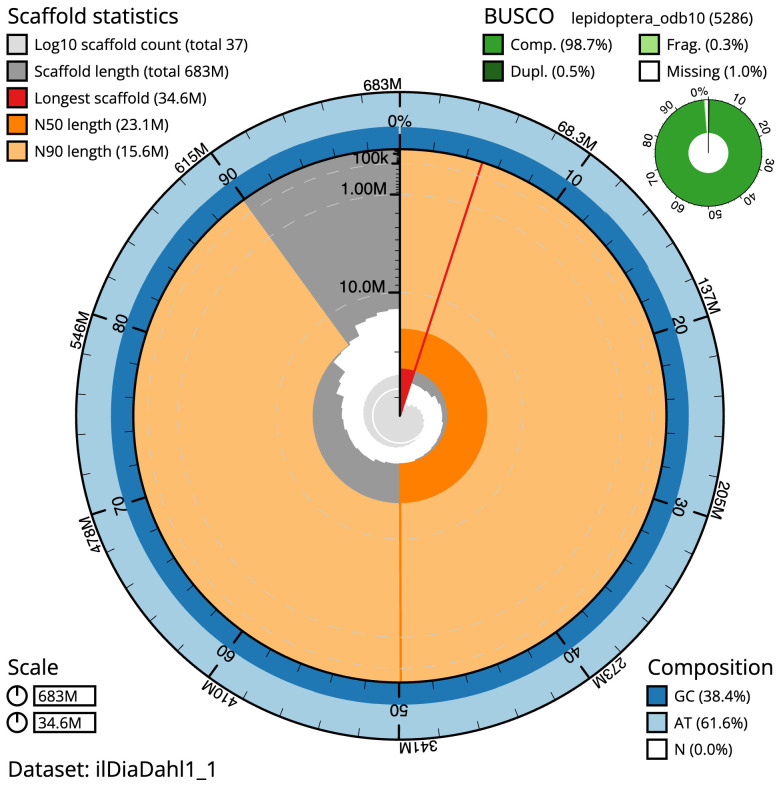 Figure 2
Figure 2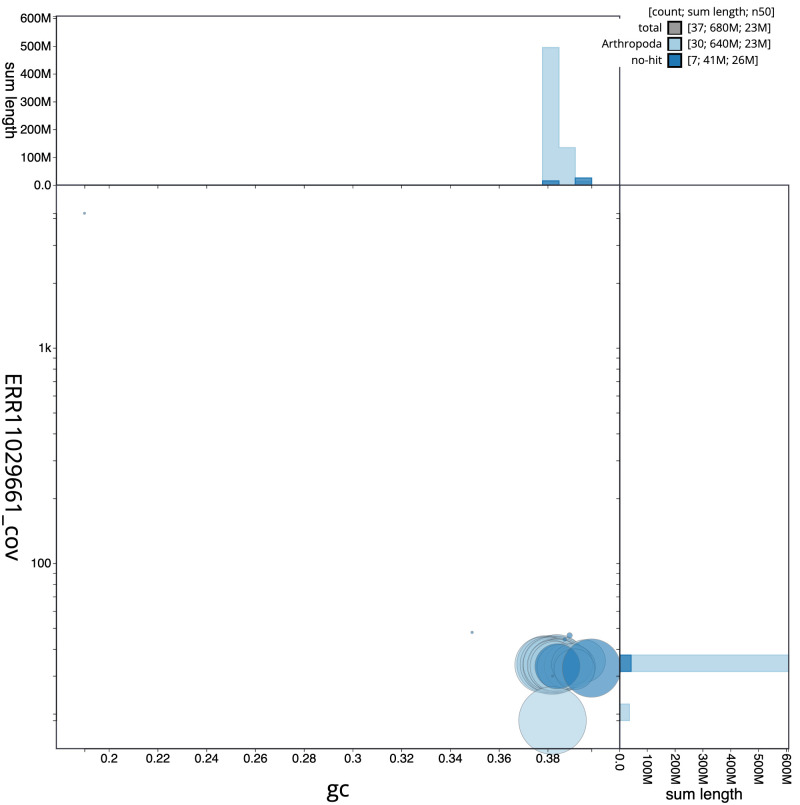 Figure 3
Figure 3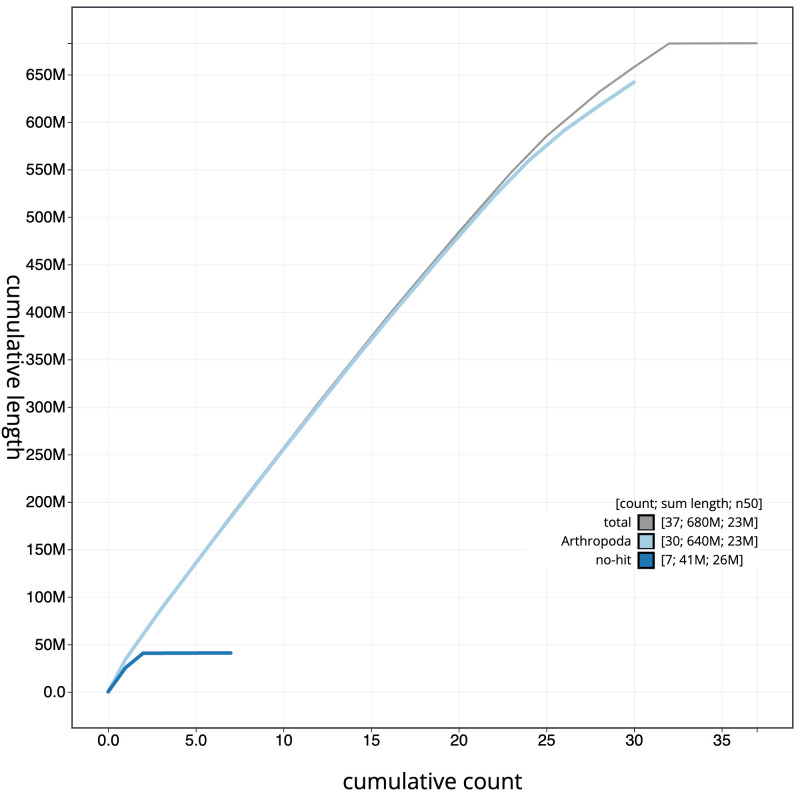 Figure 4
Figure 4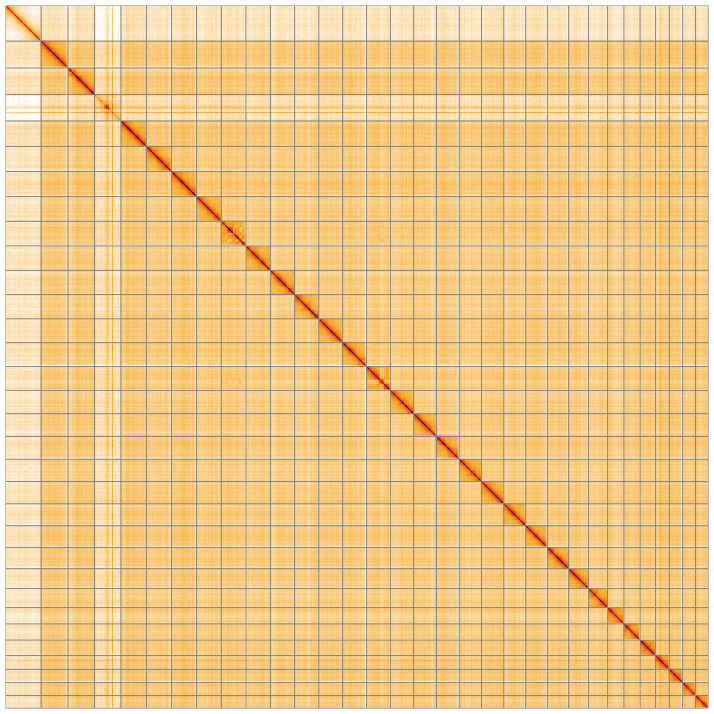 Figure 5
Figure 5| Project accession data | ||
|---|---|---|
| Assembly identifier | ilDiaDahl1.1 | |
| Species |
| |
| Specimen | ilDiaDahl1 | |
| NCBI taxonomy ID | 1804828 | |
| BioProject | PRJEB60641 | |
| BioSample and isolate information | PacBio HiFi sequencing: ilDiaDahl1, female, head and thorax SAMEA14448499
| |
| Assembly metrics
|
| |
| Consensus quality (QV) | 65.4 |
|
|
| 100.0% |
|
| BUSCO
| C:98.7%[S:98.1%,D:0.5%],F:0.3%,M:1.0%,n:5,286 |
|
| Percentage of assembly mapped to chromosomes | 99.99% |
|
| Sex chromosomes | ZW |
|
| Organelles | Mitochondrial genome: 15.36 kb |
|
| Raw data accessions | ||
| PacificBiosciences SEQUEL II | ERR11029661 | |
| Hi-C Illumina | ERR11040173 | |
| PolyA RNA-Seq Illumina | ERR11641131 | |
| Genome assembly | ||
| Assembly accession | GCA_949775195.1 | |
|
| GCA_949775585.1 | |
| Span (Mb) | 683.0 | |
| Number of contigs | 111 | |
| Contig N50 length (Mb) | 10.1 | |
| Number of scaffolds | 36 | |
| Scaffold N50 length (Mb) | 23.1 | |
| Longest scaffold (Mb) | 34.58 | |
|
| ||
| Number of protein-coding genes | 13,177 | |
| Number of non-coding genes | 2,604 | |
| Number of gene transcripts | 23,666 | |
| INSDC accession | Chromosome | Length (Mb) | GC% |
|---|---|---|---|
| 1 | 26.04 | 38.0 | |
| 2 | 25.94 | 38.5 | |
| 3 | 24.75 | 38.5 | |
| 4 | 24.38 | 38.0 | |
| 5 | 24.32 | 38.5 | |
| 6 | 24.03 | 38.0 | |
| 7 | 23.91 | 38.5 | |
| 8 | 23.77 | 38.0 | |
| 9 | 23.6 | 38.0 | |
| 10 | 23.49 | 38.0 | |
| 11 | 23.21 | 38.0 | |
| 12 | 23.14 | 38.0 | |
| 13 | 23.1 | 38.5 | |
| 14 | 22.77 | 38.0 | |
| 15 | 22.13 | 38.0 | |
| 16 | 22.0 | 38.5 | |
| 17 | 21.72 | 38.0 | |
| 18 | 21.68 | 38.5 | |
| 19 | 21.26 | 38.5 | |
| 20 | 21.01 | 38.5 | |
| 21 | 20.78 | 38.5 | |
| 22 | 19.18 | 38.0 | |
| 23 | 18.64 | 38.5 | |
| 24 | 15.83 | 39.0 | |
| 25 | 15.64 | 38.5 | |
| 26 | 15.14 | 38.5 | |
| 27 | 13.3 | 39.5 | |
| 28 | 12.99 | 39.0 | |
| 29 | 12.52 | 39.0 | |
| 30 | 12.3 | 39.0 | |
| W | 25.58 | 40.0 | |
| Z | 34.58 | 38.0 | |
| MT | 0.02 | 19.0 |
| Software tool | Version | Source |
|---|---|---|
| BlobToolKit | 4.2.1 |
|
| BUSCO | 5.3.2 |
|
| Hifiasm | 0.16.1-r375 |
|
| HiGlass | 1.11.6 |
|
| Merqury | MerquryFK |
|
| MitoHiFi | 3 |
|
| PretextView | 0.2 |
|
| purge_dups | 1.2.5 |
|
| sanger-tol/genomenote | v1.0 |
|
| sanger-tol/readmapping | 1.1.0 |
|
| YaHS | 1.2a |
|
- —Wellcome Trust
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant and Fungal Interactions Research · Insect-Plant Interactions and Control · Lepidoptera: Biology and Taxonomy
Species taxonomy
Eukaryota; Opisthokonta; Metazoa; Eumetazoa; Bilateria; Protostomia; Ecdysozoa; Panarthropoda; Arthropoda; Mandibulata; Pancrustacea; Hexapoda; Insecta; Dicondylia; Pterygota; Neoptera; Endopterygota; Amphiesmenoptera; Lepidoptera; Glossata; Neolepidoptera; Heteroneura; Ditrysia; Obtectomera; Noctuoidea; Noctuidae; Diarsia; Diarsia dahlii (Hübner, 1813) (NCBI:txid1804828).
Background
Diarsia dahlii (Hübner, 1813) also known as the Barred Chestnut ( Waring et al., 2017), is a moth in the family Noctuidae with a wingspan of 32–44 mm and 15–18 mm forewing length ( Bretherton et al., 1979; Waring et al., 2017). It is the type species of Diarsia Hübner, [1821] ( Varga & Ronkay, 2007). The moth is pale brownish or reddish with paler yellow reniform and orbicular stigmata (the latter subtended by a black dot) and contrasting slightly darker or greyish transverse fasciae or ‘bars’, especially towards the termen; compared to related species of Diarsia (‘Clays’) the forewing costa tends to be more rounded, and the species is more strongly sexually dimorphic (females more reddish in England, more purplish-blackish in Scotland, the latter confusable with D. brunnea ([Denis & Schiffermüller], 1775) ( Bretherton et al., 1979; Waring et al., 2017).
D. dahlii is univoltine, flying from mid-July to late September with no sign of a recent shift in phenology ( Randle et al., 2019), preferring woodlands, moorlands and heaths on acid soils in Britain and Ireland, where it is widespread but localised, much more so in Ireland and the south of England (e.g. woodland on greensand Surrey). It is also widespread but rather localised in Western Europe, not in the Iberian Peninsula, but much commoner in Scandinavia including the far north, with scattered records through Asia as far east as Kamchatka and Japan ( GBIF Secretariat, 2024).
In Britain and Ireland, the Barred Chestnut has declined markedly in distribution especially in the south, gone from many sites pre-1970 (with a long-term distribution decline from 1970–2016 of 30%), but the overall abundance has increased by 156% ( Randle et al. 2019).
The rounded pale whitish egg is often laid in batches and the nocturnally active larva (see e.g. Lepiforum, 2024) feeds from October to May on woody (including sallows and birches) and herbaceous (e.g. docks Rumex spp.) plants and overwinters; fullfed it is c. 34 mm long ( Bretherton et al., 1979). The shiny brown pupa is enclosed in a loose cocoon in the ground. The nocturnal adult nectars on flowers such as Calluna vulgaris (L.) Hull. and wood sage Teucrium scorodonia L. ( Bretherton et al., 1979, Lepiforum, 2024).
The mitogenome from the genomic assembly (OX459221.1) (NHMUK014451629) is at least 0.15% divergent from other DNA barcode records from England, Scotland and Norway on BOLD (11/04/2024), belonging to the BIN cluster BOLD:AAE0664, whereas it is 2.01–2.61% divergent from others in BIN BOLD:ABX6544 from Finland, Russia and Bavaria. Only one BIN is so far known in Britain, which is just 1.92% divergent from BOLD:ABX6544 (a specimen from Finland identified as D. brunnea but representing the other BIN of D. dahlii). Members of the supposed subspecies D. dahlii tibetica Boursin, 1954 from China and D. dahlii nana (Staudinger, 1892) ( Varga & Ronkay, 2007) have not yet been DNA barcoded. D. dahlii belongs to the dahlii morphological species group that includes also D. protodahlii Varga & Ronkay, 2007. The Nearctic D. esurialis (Grote, 1881) (BOLD:ABX6710) is 2.67% pairwise divergent from the haplotype from the genomic assembly; the latter species belongs to a Nearctic species group comprising also D. calgary (Smith, 1898) ( Varga & Ronkay, 2007).
The genome will be helpful in exploring sister relationships in Diarsia and examining any genomic or biological correlates of the two mitochondrial BIN clusters.
Genome sequence report
The genome was sequenced from a female Diarsia dahlii ( Figure 1) collected from United Beinn Eighe National Nature Reserve, Scotland, UK (57.63, –5.35). A total of 34-fold coverage in Pacific Biosciences single-molecule HiFi long reads was generated. Primary assembly contigs were scaffolded with chromosome conformation Hi-C data. Manual assembly curation corrected 35 missing joins or mis-joins and removed 8 haplotypic duplications, reducing the assembly length by 2.20% and the scaffold number by 24.49%, and increasing the scaffold N50 by 0.20%.
Photograph of the Diarsia dahlii (ilDiaDahl1) specimen used for genome sequencing.
The final assembly has a total length of 683.0 Mb in 36 sequence scaffolds with a scaffold N50 of 23.1 Mb ( Table 1). The snail plot in Figure 2 provides a summary of the assembly statistics, while the distribution of assembly scaffolds on GC proportion and coverage is shown in Figure 3. The cumulative assembly plot in Figure 4 shows curves for subsets of scaffolds assigned to different phyla. Most (99.99%) of the assembly sequence was assigned to 32 chromosomal-level scaffolds, representing 30 autosomes and the Z and W sex chromosomes. Chromosome-scale scaffolds confirmed by the Hi-C data are named in order of size ( Figure 5; Table 2). The Z chromosome was identified by coverage and alignment to Diarsia rubi (GCA_932274075.1) ( Boyes et al., 2023), and the W chromosome identified by read coverage. While not fully phased, the assembly deposited is of one haplotype. Contigs corresponding to the second haplotype have also been deposited. The mitochondrial genome was also assembled and can be found as a contig within the multifasta file of the genome submission.
Table 1.: Genome data for Diarsia dahlii, ilDiaDahl1.1.
Genome assembly of Diarsia dahlii, ilDiaDahl1.1: metrics.The BlobToolKit snail plot shows N50 metrics and BUSCO gene completeness. The main plot is divided into 1,000 size-ordered bins around the circumference with each bin representing 0.1% of the 682,983,932 bp assembly. The distribution of scaffold lengths is shown in dark grey with the plot radius scaled to the longest scaffold present in the assembly (34,578,549 bp, shown in red). Orange and pale-orange arcs show the N50 and N90 scaffold lengths (23,141,363 and 15,636,754 bp), respectively. The pale grey spiral shows the cumulative scaffold count on a log scale with white scale lines showing successive orders of magnitude. The blue and pale-blue area around the outside of the plot shows the distribution of GC, AT and N percentages in the same bins as the inner plot. A summary of complete, fragmented, duplicated and missing BUSCO genes in the lepidoptera_odb10 set is shown in the top right. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/ilDiaDahl1_1/dataset/ilDiaDahl1_1/snail.
Genome assembly of Diarsia dahlii, ilDiaDahl1.1: BlobToolKit GC-coverage plot.Sequences are coloured by phylum. Circles are sized in proportion to sequence length. Histograms show the distribution of sequence length sum along each axis. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/ilDiaDahl1_1/dataset/ilDiaDahl1_1/blob.
Genome assembly of Diarsia dahlii, ilDiaDahl1.1: BlobToolKit cumulative sequence plot.The grey line shows cumulative length for all sequences. Coloured lines show cumulative lengths of sequences assigned to each phylum using the buscogenes taxrule. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/ilDiaDahl1_1/dataset/ilDiaDahl1_1/cumulative.
Genome assembly of Diarsia dahlii, ilDiaDahl1.1: Hi-C contact map of the ilDiaDahl1.1 assembly, visualised using HiGlass.Chromosomes are shown in order of size from left to right and top to bottom. An interactive version of this figure may be viewed at https://genome-note-higlass.tol.sanger.ac.uk/l/?d=cC69O4MsTNqCZ3Q0Wd-WZQ.
Table 2.: Chromosomal pseudomolecules in the genome assembly of Diarsia dahlii, ilDiaDahl1.
The estimated Quality Value (QV) of the final assembly is 65.4 with k-mer completeness of 100.0%, and the assembly has a BUSCO v5.3.2 completeness of 98.7% (single = 98.1%, duplicated = 0.5%), using the lepidoptera_odb10 reference set ( n = 5,286).
Metadata for specimens, barcode results, spectra estimates, sequencing runs, contaminants and pre-curation assembly statistics are given at https://links.tol.sanger.ac.uk/species/1804828.
Genome annotation report
The Diarsia dahlii genome assembly (GCA_949775195.1) was annotated at the European Bioinformatics Institute (EBI) on Ensembl Rapid Release. The resulting annotation includes 23,666 transcribed mRNAs from 13,177 protein-coding and 2,604 non-coding genes ( Table 1; https://rapid.ensembl.org/Diarsia_dahlii_GCA_949775195.1/Info/Index).
Methods
Sample acquisition and nucleic acid extraction
A female Diarsia dahlii (specimen ID NHMUK014451629, ToLID ilDiaDahl1) was collected from Beinn Eighe National Nature Reserve, Scotland, UK (latitude 57.63, longitude –5.35) on 2021-09-09 using a light trap. The specimen was collected and identified by David Lees (Natural History Museum) and preserved by dry freezing at –80 °C.
The workflow for high molecular weight (HMW) DNA extraction at the Wellcome Sanger Institute (WSI) includes a sequence of core procedures: sample preparation; sample homogenisation, DNA extraction, fragmentation, and clean-up. In sample preparation, the ilDiaDahl1 sample was weighed and dissected on dry ice ( Jay et al., 2023). Tissue from the head and thorax was homogenised using a PowerMasher II tissue disruptor ( Denton et al., 2023a).
HMW DNA was extracted in the WSI Scientific Operations core using the Automated MagAttract v2 protocol ( Oatley et al., 2023). The DNA was sheared into an average fragment size of 12–20 kb in a Megaruptor 3 system with speed setting 31 ( Bates et al., 2023). Sheared DNA was purified by solid-phase reversible immobilisation ( Strickland et al., 2023): in brief, the method employs a 1.8X ratio of AMPure PB beads to sample to eliminate shorter fragments and concentrate the DNA. The concentration of the sheared and purified DNA was assessed using a Nanodrop spectrophotometer and Qubit Fluorometer and Qubit dsDNA High Sensitivity Assay kit. Fragment size distribution was evaluated by running the sample on the FemtoPulse system.
RNA was extracted from remaining head and thorax tissue of ilDiaDahl1 in the Tree of Life Laboratory at the WSI using the RNA Extraction: Automated MagMax™ mirVana protocol ( do Amaral et al., 2023). The RNA concentration was assessed using a Nanodrop spectrophotometer and a Qubit Fluorometer using the Qubit RNA Broad-Range Assay kit. Analysis of the integrity of the RNA was done using the Agilent RNA 6000 Pico Kit and Eukaryotic Total RNA assay.
Protocols developed by the WSI Tree of Life laboratory are publicly available on protocols.io ( Denton et al., 2023b).
Sequencing
Pacific Biosciences HiFi circular consensus DNA sequencing libraries were constructed according to the manufacturers’ instructions. Poly(A) RNA-Seq libraries were constructed using the NEB Ultra II RNA Library Prep kit. DNA and RNA sequencing was performed by the Scientific Operations core at the WSI on Pacific Biosciences SEQUEL II (HiFi) and Illumina NovaSeq 6000 (RNA-Seq) instruments. Hi-C data were also generated from abdomen tissue of ilDiaDahl1 using the Arima v2 kit. The Hi-C sequencing was performed using paired-end sequencing with a read length of 150 bp on the Illumina NovaSeq 6000 instrument.
Genome assembly, curation and evaluation
Assembly was carried out with Hifiasm ( Cheng et al., 2021) and haplotypic duplication was identified and removed with purge_dups ( Guan et al., 2020). The assembly was then scaffolded with Hi-C data ( Rao et al., 2014) using YaHS ( Zhou et al., 2023). The assembly was checked for contamination and corrected as described previously ( Howe et al., 2021). Manual curation was performed using HiGlass ( Kerpedjiev et al., 2018) and PretextView ( Harry, 2022). The mitochondrial genome was assembled using MitoHiFi ( Uliano-Silva et al., 2023), which runs MitoFinder ( Allio et al., 2020) or MITOS ( Bernt et al., 2013) and uses these annotations to select the final mitochondrial contig and to ensure the general quality of the sequence.
A Hi-C map for the final assembly was produced using bwa-mem2 ( Vasimuddin et al., 2019) in the Cooler file format ( Abdennur & Mirny, 2020). To assess the assembly metrics, the k-mer completeness and QV consensus quality values were calculated in Merqury ( Rhie et al., 2020). This work was done using Nextflow ( Di Tommaso et al., 2017) DSL2 pipelines “sanger-tol/readmapping” ( Surana et al., 2023a) and “sanger-tol/genomenote” ( Surana et al., 2023b). The genome was analysed within the BlobToolKit environment ( Challis et al., 2020) and BUSCO scores ( Manni et al., 2021; Simão et al., 2015) were calculated.
Table 3 contains a list of relevant software tool versions and sources.
Genome annotation
The Ensembl Genebuild annotation system ( Aken et al., 2016) was used to generate annotation for the Diarsia dahlii assembly (GCA_949775195.1) in Ensembl Rapid Release at the EBI. Annotation was created primarily through alignment of transcriptomic data to the genome, with gap filling via protein-to-genome alignments of a select set of proteins from UniProt ( UniProt Consortium, 2019).
Wellcome Sanger Institute – Legal and Governance
The materials that have contributed to this genome note have been supplied by a Darwin Tree of Life Partner. The submission of materials by a Darwin Tree of Life Partner is subject to the ‘Darwin Tree of Life Project Sampling Code of Practice’, which can be found in full on the Darwin Tree of Life website here. By agreeing with and signing up to the Sampling Code of Practice, the Darwin Tree of Life Partner agrees they will meet the legal and ethical requirements and standards set out within this document in respect of all samples acquired for, and supplied to, the Darwin Tree of Life Project.
Further, the Wellcome Sanger Institute employs a process whereby due diligence is carried out proportionate to the nature of the materials themselves, and the circumstances under which they have been/are to be collected and provided for use. The purpose of this is to address and mitigate any potential legal and/or ethical implications of receipt and use of the materials as part of the research project, and to ensure that in doing so we align with best practice wherever possible. The overarching areas of consideration are:
• Ethical review of provenance and sourcing of the material
• Legality of collection, transfer and use (national and international)
Each transfer of samples is further undertaken according to a Research Collaboration Agreement or Material Transfer Agreement entered into by the Darwin Tree of Life Partner, Genome Research Limited (operating as the Wellcome Sanger Institute), and in some circumstances other Darwin Tree of Life collaborators.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdennur N Mirny LA : Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36(1):311–316. 10.1093/bioinformatics/btz 540 31290943 PMC 8205516 · doi ↗ · pubmed ↗
- 2Aken BL Ayling S Barrell D : The Ensembl gene annotation system. Database (Oxford). 2016;2016: baw 093. 10.1093/database/baw 093 27337980 PMC 4919035 · doi ↗ · pubmed ↗
- 3Allio R Schomaker-Bastos A Romiguier J : Mito Finder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020;20(4):892–905. 10.1111/1755-0998.13160 32243090 PMC 7497042 · doi ↗ · pubmed ↗
- 4Bates A Clayton-Lucey I Howard C : Sanger Tree of Life HMW DNA fragmentation: diagenode Megaruptor®3 for LI Pac Bio. protocols.io. 2023. 10.17504/protocols.io.81wgbxzq 3lpk/v 1 · doi ↗
- 5Bernt M Donath A Jühling F : MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69(2):313–319. 10.1016/j.ympev.2012.08.023 22982435 · doi ↗ · pubmed ↗
- 6Boyes D Holland PWH , University of Oxford and Wytham Woods Genome Acquisition Lab, : The genome sequence of the small square-spot, Diarsia rubi (Vieweg, 1790) [version 1; peer review: awaiting peer review]. Wellcome Open Res. 2023;8:210. 10.12688/wellcomeopenres.19299.1 39211527 PMC 11358679 · doi ↗ · pubmed ↗
- 7Bretherton RF Goater B Lorimer RI : Noctuidae: noctuinae and hadeninae.In: Heath, J. and Emmet, A. M. (eds.) The Moths and Butterflies of Great Britain & Ireland.London: Curwen Books,1979;9:120–278. 10.1163/9789004627161_012 · doi ↗
- 8Challis R Richards E Rajan J : Blob Tool Kit - interactive quality assessment of genome assemblies. G 3 (Bethesda). 2020;10(4):1361–1374. 10.1534/g 3.119.400908 32071071 PMC 7144090 · doi ↗ · pubmed ↗