Uncommon Etiology of Pancreatic Mass: a Case Report
Sanae Moqran, Layla Tahiri El Ousrouti, Nawal Hammas, Nizar El Bouardi, Laila Chbani

TL;DR
This case report highlights how IgG4-related autoimmune pancreatitis can mimic pancreatic cancer, emphasizing the need for accurate diagnosis to avoid unnecessary surgery.
Contribution
The paper presents a clinical case underscoring the diagnostic challenges between IgG4-related AIP and pancreatic cancer.
Findings
IgG4-related AIP can clinically and radiologically resemble pancreatic cancer.
Histopathological and immunohistochemical analysis is essential for accurate diagnosis.
Misdiagnosis can lead to unnecessary surgical interventions.
Abstract
IgG4-related autoimmune pancreatitis (AIP) is a chronic inflammatory disease of the pancreas with a distinct histological feature. Its diagnosis remains challenging as some features overlap with pancreatic cancer. We present a case of IgG4-related AIP mimicking pancreatic cancer. A 70-year-old male patient presented with epigastric pain, radiating to the entire abdomen with an unquantified weight loss. Magnetic resonance cholangiopancreatography (MRCP) showed a mass with a 28 mm long axis, in the head of the pancreas with pancreatic duct dilatation. Thus, it was presumed to be a pancreatic neoplasm and pancreatic resection was undertaken without a definitive preoperative diagnosis. In terms of clinical presentation, imaging characteristics, and laboratory parameters, IgG4-related AIP can resemble pancreatic cancer. Thus, histopathological studies remain the gold standard for a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
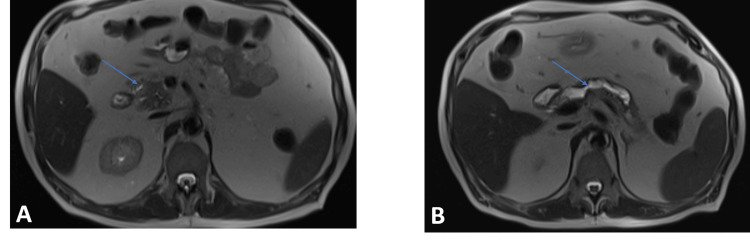 Figure 1
Figure 1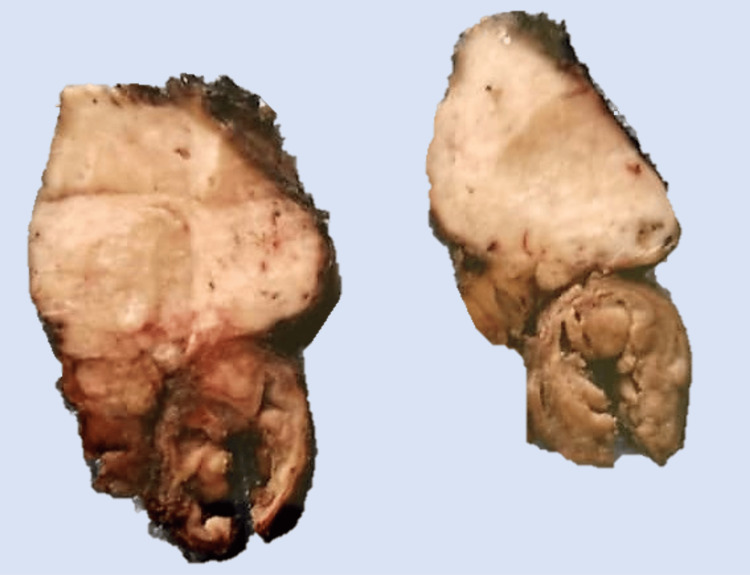 Figure 2
Figure 2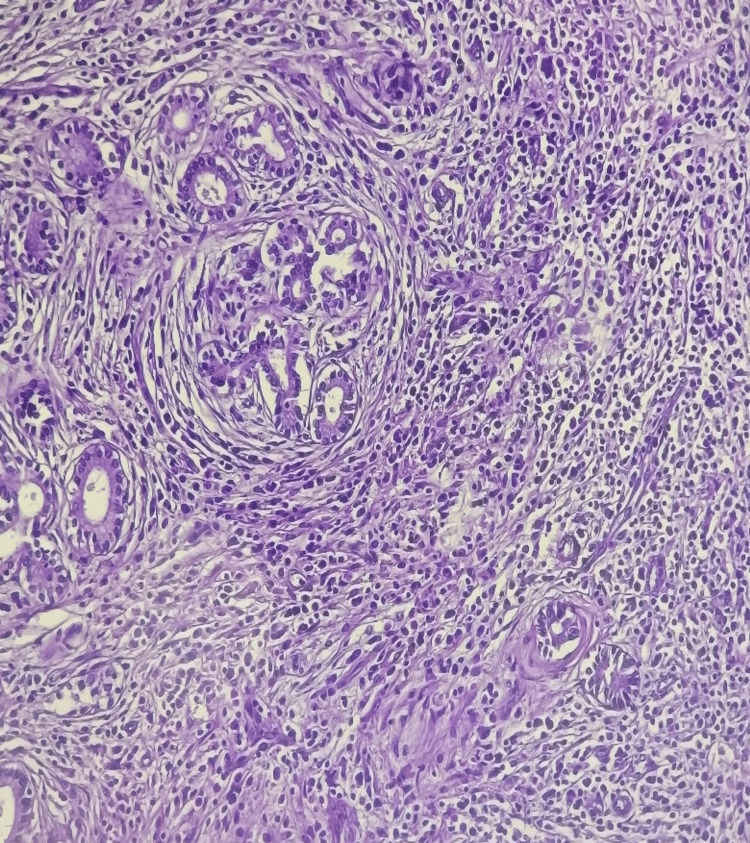 Figure 3
Figure 3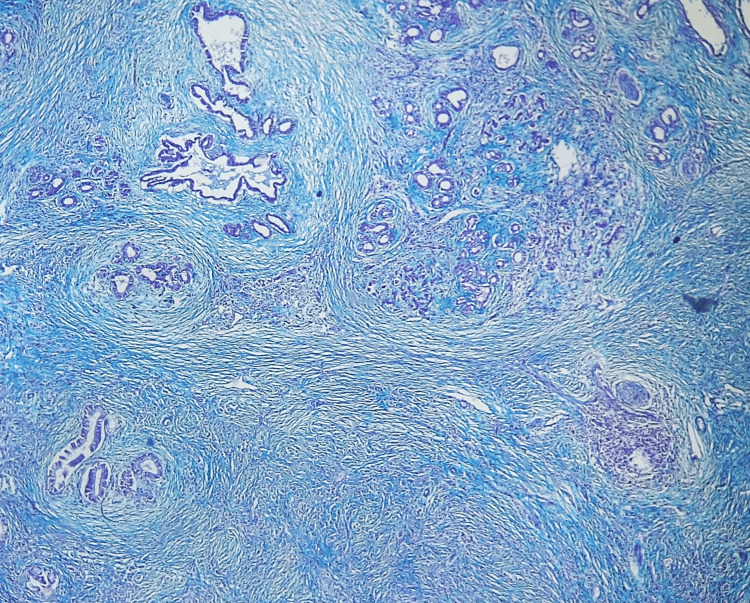 Figure 4
Figure 4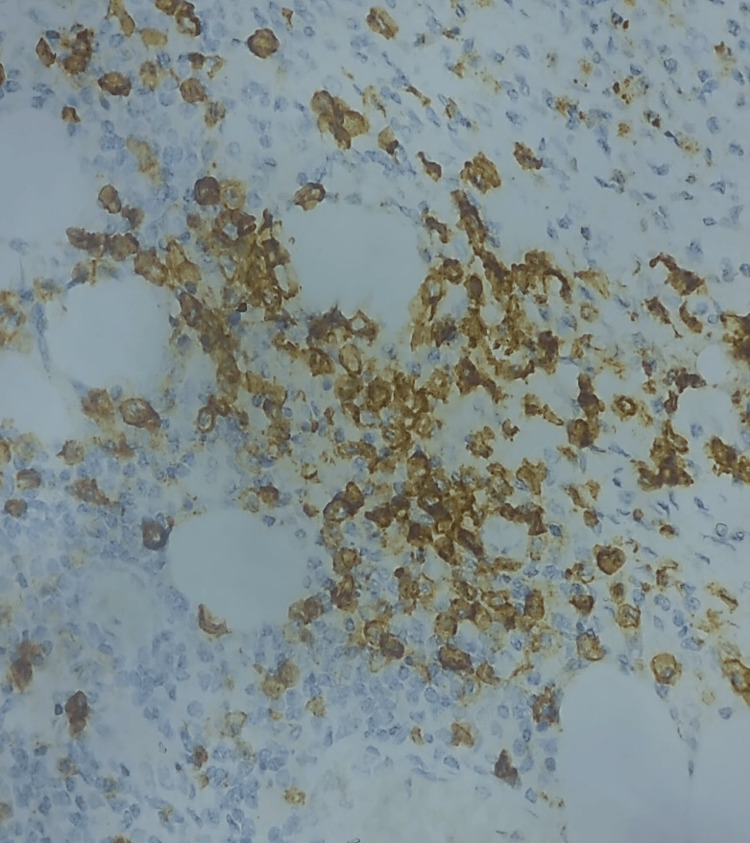 Figure 5
Figure 5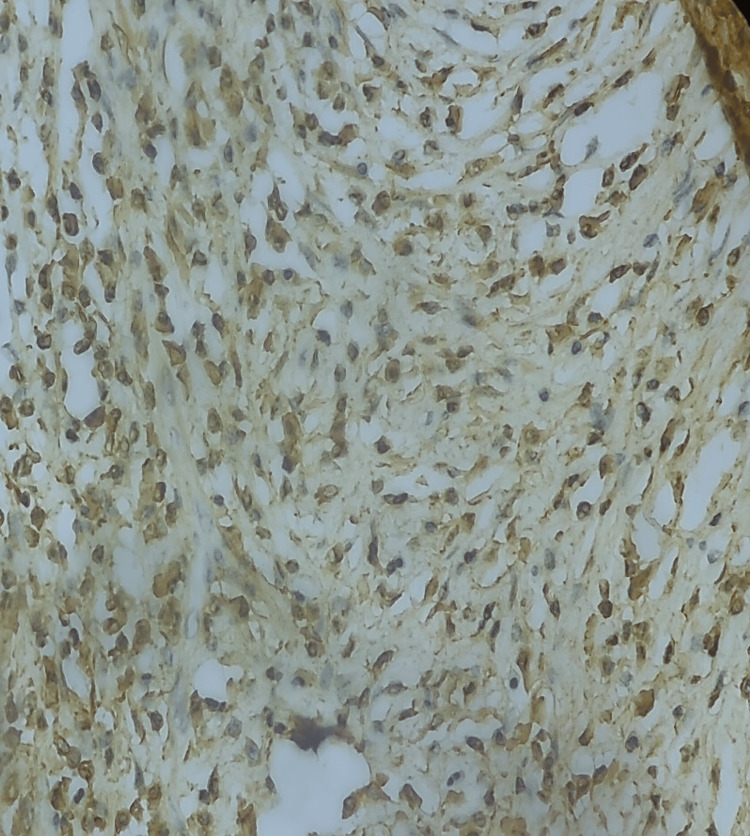 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIgG4-Related and Inflammatory Diseases · Neuroendocrine Tumor Research Advances · Gastrointestinal disorders and treatments
Introduction
Autoimmune pancreatitis (AIP) is an uncommon form of chronic pancreatitis caused by an immunological reaction, resulting in inflammation in the pancreas. Two types of AIP exist: type 1 or IgG4-related AIP, and type 2, which is also known as idiopathic duct-centric pancreatitis, a disease unrelated to IgG4. Type 1 AIP is characterized by pancreatic symptoms that occur within a broader systemic fibroinflammatory disorder, known as IgG4-associated, which can affect various organs in the body [1].
It should be noted that there are overlapping clinical and radiological symptoms between the two conditions. A precise diagnosis requires a combination of clinical, radiological, serological, and histopathological examination. The main differential diagnosis is pancreatic cancer, underscoring the paramount need for an accurate diagnosis. Accurate diagnosis is crucial as steroid medication has been demonstrated to be highly effective, and surgical intervention is redundant in most cases.
This article was previously presented as a poster at the 35th European Congress of Pathology in September 2023 and subsequently published as an abstract in the Virshow archive.
Case presentation
A 70-year-old male patient, who was undergoing hemodialysis for end-stage renal failure and had a chronic interstitial lung disease associated with fibrosis, presented with epigastric pain radiating to his whole abdomen and unquantified weight loss. No jaundice was observed. The patient's test results lacked specificity, with bilirubin at 4.6, glutamic oxaloacetic transaminase (GOT)/glutamic pyruvic transaminase (GPT) at 25/11.3, alkaline phosphatase (ALP)/gamma-glutamyl transferase (GGT) at 80/77, and tumour marker values of CA-19 at 65.6 U/mL (reference range, 0-35 U/mL). Magnetic resonance cholangiopancreatography (MRCP) revealed a 28 mm mass in the pancreatic head, causing upstream main pancreatic duct dilatation (Figure 1).
Presence of a cephalic pancreatitis mass (A) resulting in the dilation of the upstream Wirsung duct (B)
Based on these findings, the presence of a pancreatic neoplasm was suspected, and the patient proceeded to undergo pancreatic resection without a clear preoperative diagnosis. On gross evaluation (Figure 2), a solid and white mass was found in the head of the pancreas.
Gross evaluation showing a firm and white unlimited mass in the head of pancreas
On histological examination, it was revealed that the pancreatic parenchyma was largely separated by storiform collagen bundles and accompanied by a dense, inflammatory infiltrate that contained a high number of plasma cells (Figures 3, 4).
Diffuse lymphoplasmacytic infiltration of the pancreas and marked storiform fibrosis.
Storiform fibrosis more discernible after Masson’s trichrome staining.
Based on the results, AIP IgG4-related diagnosis was suspected and additional immunohistochemical tests were required to confirm the diagnosis. On immunohistochemistry, the anti-CD138 antibody verified the prevalence of plasma cells, most of which were positive for IgG4 (Figures 5, 6).
Highly magnified view showing diffuse infiltration by CD138-positive plasma cells.
IgG4-positive plasma cells seen in abundance in the pancreas.
Discussion
IgG4-related AIP represents the pancreatic manifestation of IgG4-associated disease, which is a systemic fibroinflammatory disorder. The first report of this condition dates back to 1892, when Muckilicz described a patient with symmetrical enlargement of salivary glands and extensive mononuclear cell infiltration [1]. In 1991, Kawaguchi identified the histological features of pancreatic AIP, naming it lymphoplasmacytic sclerosing pancreatitis (LPSP), four years prior to Yoshida's proposal of the term PAI [2]. It took Hamano another decade to discover the association between elevated IgG4 serum levels and LPSP [3]. Studies subsequent to this publication provided evidence of the infiltration of inflammatory tissues with plasma cells containing IgG4 antibodies in the pancreas of LPSP patients [4]. In 2003, a Japanese team reported the phrase 'IgG4-related AIP' as a novel clinicopathological entity within the wider multifocal disorder of IgG4-associated diseases [5].
Due to the novelty of this diagnosis and lack of recognition, there is limited understanding of the epidemiology of this disease. Japanese researchers showed great interest in studying this recently discovered entity. The Japanese National Epidemiological Survey indicated a prevalence of 4.6/100000 individuals and an incidence of 1.4 per 100,000. It is more probable to affect men than women. The Japanese survey reported a mean patient age of 68.1 years [6], reminiscent of pancreatic cancer's demographic profile.
IgG4-related AIP presents commonly with painless obstructive jaundice resulting from biliary obstruction, in addition to non-specific symptoms such as nausea, vomiting or weight loss [7]. Laboratory tests are not specific and only elevated serum IgG4 levels have been described in numerous series, with sensitivity and specificity ranging from 67% to 94% and 89% to 100%, respectively, according to a meta-analysis of seven studies [8].
Abdominal computed tomography (CT) and magnetic resonance imaging (MRI) are frequently employed to assess alterations in the pancreatic parenchyma. In the context of AIP, these imaging techniques demonstrate either diffuse hypertrophy or focal hypertrophy of the pancreas, contingent on the extent of the pathology. In cases of focal hypertrophy, pancreatic cancer should be considered as a primary diagnosis in order to exclude it. In the presented case, AIP and pancreatic cancer share numerous clinical and radiological features, including a pancreatic mass and dilatation of the main upstream pancreatic duct.
Upon gross evaluation, the disease predominantly affects the pancreatic head. On occasion, the entire gland is affected as well. The pancreas exhibits a firm to hard consistency. The presence of a discernible mass is infrequent, posing a diagnostic challenge for malignancy [9].
Histology remains a critical component of the diagnosis of AIP type 1. Extensive lymphoplasmacytic infiltration rich in plasma cells, fibrosis at least focally arranged in a storiform pattern, and obliterative phlebitis are the three key histological findings mentioned. Two other features that have been reported in the literature but are neither sensitive nor specific for diagnosis are phlebitis without luminal obliteration and an increase in eosinophils [10]. Immunohistochemistry shows a predominance of IgG4-positive plasma cells. A number of more than 50 IgG4-positive plasma cells per high-power field (HPF) has been described in the literature and appears to be specific, as it has never been described in any other pancreatic pathology [11-15]. It should be emphasised that histological and anatomopathological evaluations are rarely available prior to surgical resection, as most surgical teams do not perform a biopsy in the case of pseudotumours suggesting malignancy. This may account for the 5-10% incorrect surgical procedures reported in the literature [16,17].
Given the lack of specificity of the clinical and even radiological features of AIP in particular and IgG4 disease in general, the diagnosis of IgG4-related AIP can sometimes be a real challenge, requiring the collection of a number of arguments. Many guidelines have been proposed for the diagnosis of igG4-related disease; the most commonly used criteria in the literature are those proposed by a Japanese consensus, called "Comprehensive Diagnostic Criteria" or CDC [18]. These are general criteria that are based on a combination of clinical, biological, and histological criteria and are used to categorize patients as having "possible" IgG4-related disease (in the presence of clinical and biological criteria), "probable" (in the presence of clinical and histological criteria) or "definite" (in the presence of the three criteria) [18]. A lack of biopsy samples meant that these criteria had relatively low sensitivity in individuals with IgG4-related AIP [19]. The identification of additional diagnostic criteria, such as the 2019 ACR/EULAR IgG4-RD classification [20], has therefore proven crucial in addressing the challenges posed by ambiguous cases. One of the defining characteristics of this diagnostic criteria set is that a patient can be reliably identified as having IgG4-RD even in the absence of a biopsy or an elevated serum IgG4 level in a significant number of cases [20]. In our case, the diagnosis of IgG4-related AIP was maintained based on histological and immunohistochemical findings.
In accordance with the aforementioned criteria, the diagnosis of AIP can be made without the need for invasive methods, provided that the presence of adequate radiological imaging and raised serum IgG4 levels can be demonstrated. Nevertheless, certain cases, such as those with a pseudotumor phenotype that initially suggests pancreatic cancer, may prove challenging to diagnose. In such cases, it is imperative to perform a pancreatic biopsy in order to prevent the implementation of an unwarranted surgical intervention.
In contrast to pancreatic adenocarcinoma, where surgical intervention is frequently indicated, simple corticosteroid therapy is typically sufficient to treat AIP. Nevertheless, immunotherapy may be necessary in the event of a relapse or resistance.
Conclusions
The diagnosis of AIP is based on a combination of clinical, biological, radiological, and anatomopathological evidence. Effective communication between the clinician, radiologist, and pathologist is therefore essential to ensure a correct diagnosis.
It is critical to distinguish pancreatic cancer from IgG4-related AIP, as the two conditions have completely different prognoses and require different treatments. A misdiagnosis as a malignancy can lead to inappropriate surgical interventions. It is therefore imperative that a biopsy be conducted prior to any invasive treatment, including surgery, given that the associated complications merit due consideration.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Diagnosis and classification of autoimmune pancreatitis Autoimmun Rev Okazaki K Tomiyama T Mitsuyama T Sumimoto K Uchida K 4514581320142442418410.1016/j.autrev.2014.01.010 · doi ↗ · pubmed ↗
- 2Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis Dig Dis Sci Yoshida K Toki F Takeuchi T 156115687199510.1007/BF 022852097628283 · doi ↗ · pubmed ↗
- 3High serum Ig G 4 concentrations in patients with sclerosing pancreatitis N Engl J Med Aithal GP Breslin NP Gumustop B 147148345200110.1056/NEJM 20010712345021511450670 · doi ↗ · pubmed ↗
- 4Ig G 4-positive plasma cells specifically infiltrate various organs in autoimmune pancreatitis Pancreas Kamisawa T 16716829200410.1097/00006676-200408000-0001415257111 · doi ↗ · pubmed ↗
- 5A new clinicopathological entity of Ig G 4-related autoimmune disease J Gastroenterol Kamisawa T Funata N Hayashi Y 9829843820031461460610.1007/s 00535-003-1175-y · doi ↗ · pubmed ↗
- 6Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2016 J Gastroenterol Masamune A Kikuta K Hamada S Tsuji I Takeyama Y Shimosegawa T Okazaki K 462470552020
- 7Clinical profile of autoimmune pancreatitis and its histological subtypes: an international multicenter survey Pancreas Kamisawa T Chari ST Giday SA 8098144020112174731010.1097/MPA.0b 013e 3182258 a 15 · doi ↗ · pubmed ↗
- 8Usefulness of serum Ig G 4 in the diagnosis and follow up of autoimmune pancreatitis: a systematic literature review and meta-analysis Gastroenterol Hepatol Morselli-Labate AM Pezzilli RJ 153624200910.1111/j.1440-1746.2008.05676.x 19067780 · doi ↗ · pubmed ↗