Exploration on cold adaptation of Antarctic lichen via detection of positive selection genes
Yanyan Wang, Yaran Zhang, Rong Li, Ben Qian, Xin Du, Xuyun Qiu, Mengmeng Chen, Guohui Shi, Jiangchun Wei, Xin-Li Wei, Qi Wu

TL;DR
This study explores how the Antarctic lichen Usnea aurantiacoatra adapts to cold by identifying genes under positive selection and testing their role in cold resistance.
Contribution
The paper identifies specific genes and protein networks in Antarctic lichen that are under positive selection and functionally linked to cold adaptation.
Findings
Positively selected genes in Antarctic lichen are enriched in transmembrane transporter activity and vacuole components.
Two protein interaction networks related to RNA helicase and G-protein signaling were identified.
Disruption of UaRgs1 increased cold sensitivity, suggesting its role in cold resistance.
Abstract
Lichen as mutualistic symbiosis is the dominant organism in various extreme terrestrial environment on Earth, however, the mechanisms of their adaptation to extreme habitats have not been fully elucidated. In this study, we chose the Antarctic dominant lichen species Usnea aurantiacoatra to generate a high-quality genome, carried out phylogenetic analysis using maximum likelihood and identify genes under positive selection. We performed functional enrichment analysis on the positively selected genes (PSGs) and found that most of the PSGs focused on transmembrane transporter activity and vacuole components. This suggest that the genes related to energy storage and transport in Antarctic U. aurantiacoatra were affected by environmental pressure. Inside of the 86 PSGs screened, two protein interaction networks were identified, which were RNA helicase related proteins and regulator of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
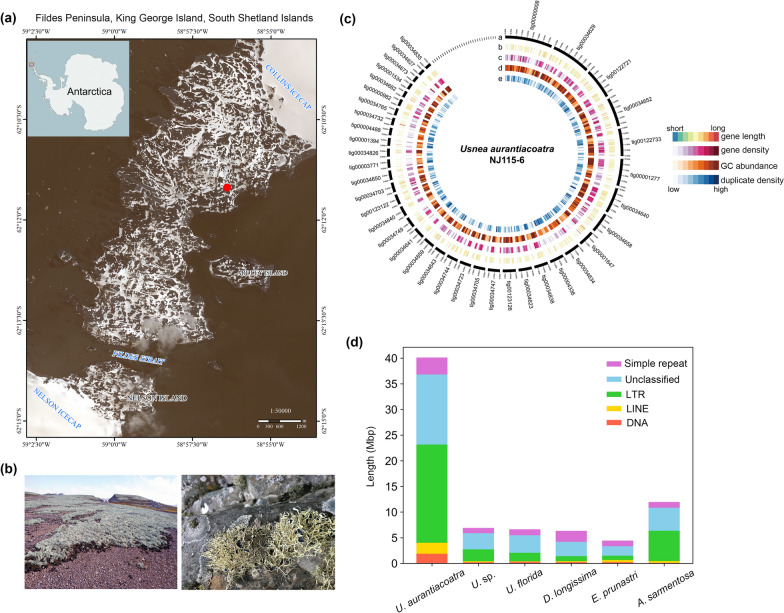 Figure 1
Figure 1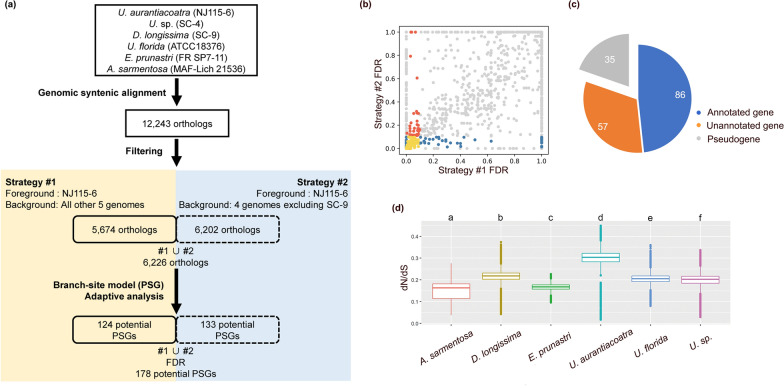 Figure 2
Figure 2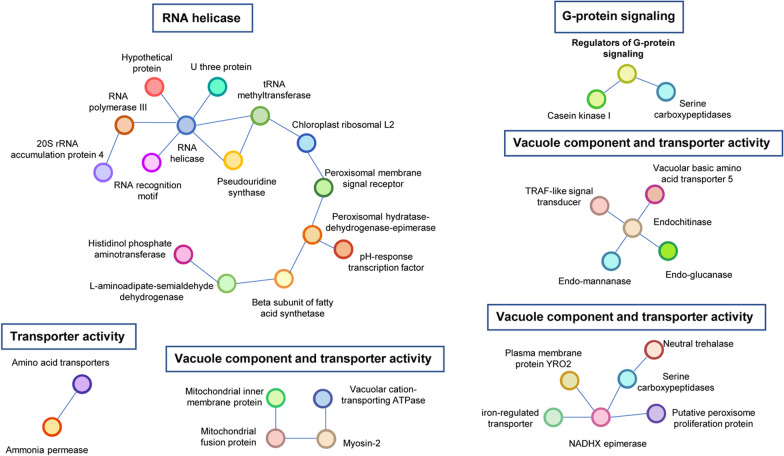 Figure 3
Figure 3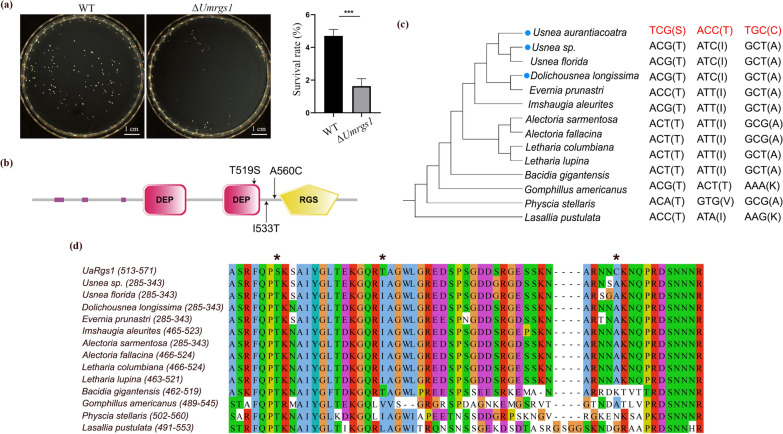 Figure 4
Figure 4- —Strategic Priority Research Program of Science
- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
- —Space Application System of China Manned Space Program
- —Strategic Priority Research Program of Science
- —http://dx.doi.org/10.13039/100014718Innovative Research Group Project of the National Natural Science Foundation of China
- —Senior User Project of RV KEXUE
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLichen and fungal ecology · Polar Research and Ecology · Botany and Plant Ecology Studies
INTRODUCTION
Lichens are stable mutualistic symbiosis composed of fungi and algae or cyanobacteria, also including a diverse microbiome (Honegger 1991, Lücking and Nelsen 2018, Hawksworth and Grube 2020; Zhang et al. 2023). In lichens, lichen-forming fungi (LFF) provide protection to algae, whereas the algae provide photosynthetic nutrition for the LFF. This symbiotic form allows lichens to be widely distributed in extreme environments worldwide, including polar, plateau, and desert regions. For this reason, lichens are known as “pioneer organisms” of the terrestrial ecosystem and stress-tolerant extremophiles (Yang et al. 2023). Taking the Antarctica as an example, it is the coldest continent in the world with 99.8% of the area covered by ice (Burton-Johnson et al. 2016). In its northern region, there are some ice-free and relatively mild sites called the Maritime Antarctica such as Fildes Peninsula of King George Island (Fig. 1a), where is characterized by annual mean temperature − 3 to − 4 °C, sometimes lower than − 10 to − 12 °C in over 8-month-long winter, and common freeze–thaw cycle. However, lichens not only can survive but also are predominant organisms with over 400 species in Antarctic vegetation here, as comparison, only two vascular plant species exist (Øvstedal and Smith 2001).Fig. 1. Antarctic Usnea aurantiacoatra and its genome annotations. a Location of the Fildes Peninsula on the Antarctic continent and the sampling site of U. aurantiacoatra. The sampling site is marked with solid red circles. b Habitat of U. aurantiacoatra in Ardley Island, which looks like a grassland from a distance. And the morphology of saxicolous lichen thallus in the field. c Genome annotations of U. aurantiacoatra (NJ115-6). Circos representation of detailed information about the genome. a: Contigs over 500 Kb are labeled with names; b: Gene length; c: gene density (gene numbers per 100 Kb); d: GC abundance (GC percentage per 100 Kb); e: duplicate density (duplicate numbers per 100 Kb). d Abundance of repetitive elements in six lichen genomes. The repetitive elements in U. aurantiacoatra was nearly 40 Mbp, which far exceeds the amount in the other five genomes
Although knowledge of the lichens has been accumulated on biodiversity and conservation (Romeike et al. 2002; Wauchope et al. 2019), as well as how they are influenced by Antarctic climate change (Sancho et al. 2017), the mechanism by which lichen species are adaptive to such an extreme environment and dominate the terrestrial vegetation is yet to be fully elucidated. The corresponding researches were only reported on animals (Li et al. 2014a; Daane and Detrich 2022) and green algae (Zhang et al. 2020) here. To our knowledge, no Antarctic lichen genome has yet been sequenced and analyzed. Moreover, the number of sequenced genomes is still too little in relation to the great variety of lichen species. The lack of enough genetic information, as well as limitations such as slow growth and difficulty in genetic manipulation, have slowed down the development of research on lichen resistance mechanism. In this context, genomics will be a more effective and feasible strategy.
Through our field investigation in the Fildes Peninsula of King George Island in Antarctica, we found the lichen species Usnea aurantiacoatra thriving there, and covering the ground gravel like grassland (Fig. 1a-1b), suggesting strong adaptation to the polar environment. Biogeographic and phylogenetic studies also indicated some special characteristics of this lichen. Firstly, majority of usneoid lichens distributed in low- and medium-latitude regions, including most species of Usnea and other related genera, whereas only several Usnea species including U. aurantiacoatra are confined in Antarctica. Secondly, these Usnea species confined in Antarctica and high-latitude represent one separate subclade, suggesting a significant genetic differentiation from the usneoid ancestors (Øvstedal and Smith 2001; Wirtz et al. 2006; Thell et al. 2012; Divakar et al. 2017). Based on these comprehensive characteristics, U. aurantiacoatra was chosen as our research model for cold adaptation of lichens.
In this study, three LFFs were de novo whole-genome sequenced, yielding a high-quality genome of U. aurantiacoatra. Genomic syntenic alignment was comprehensively performed in six usneoid LFFs, yielding a list of positively selected genes for environmental adaptation in U. aurantiacoatra. We performed functional enrichment and protein interaction network analysis and found that positively selected genes cluster into vacuole components, transporter proteins, RNA helicase, and G-protein signaling. We chose to use Umbilicaria muhlenbergii for functional validation of positively selected genes because U. muhlenbergii has genetic manipulation system (Wang et al. 2020, 2023), and it also grows extensively in cold region like U. aurantiacoatra, both of which belong to Lecanoromycetes. Protein interaction network analyses indicated that UaRGS1 may play an important role in the adaptation of U. aurantiacoatra to the Antarctic environment, and that deletion of its ortholog, UmRGS1, in U. muhlenbergii resulted in the inability of U. muhlenbergii to tolerate cold-shock. Our results provided evidence for understanding the adaptation of U. aurantiacoatra to Antarctic environments and would open up the study of adaptive mechanisms in lichens at molecular level.
METHODS
Sample information
The Usnea aurantiacoatra lichen sample (NJ115-6) in minor amount was collected from the Fildes Peninsula, King George Island in Antarctica (Fig. 1a-1b, Table S1). The other two usneoid lichens, Dolichousnea longissima (coll.no. SC-9, syn. Usnea longissima) and an unknown Usnea species (coll.no. SC-4), were collected from the Sichuan Province of China (Table S1), considering their cold habitat source due to the high altitudes, and close phylogeny to U. aurantiacoatra. All samples were identified based on morphological observation and nuclear ribosomal DNA internal transcribed spacer (ITS) sequences, which were amplified using ITS4/ITS5 (White et al. 1990), and the sequencing results were blasted in the NCBI database.
DNA extraction
The thallus of U. aurantiacoatra (NJ115-6), D. longissima (SC-9), and U. sp. (SC-4) were surface-cleaned and sterilized by immersion in 75% ethanol for 1 min, 1% sodium hypochlorite for 1 min, and sterile water for 1 min. Samples were then aired on sterile filter paper for half a day in a clean bench. The U. aurantiacoatra (NJ115-6) sample was directly sent to Nextomics Biosciences in Wuhan, China, and the thallus of D. longissima (SC-9) and that of U. sp. (SC-4) were sent to Majorbio in Shanghai, China, for DNA extraction and genomic sequencing.
De novo whole-genome sequencing
A sequencing strategy, combining second-generation, Hi-C, and third-generation sequencing techniques, was used for the sequencing of usneoid lichen genomes. After quality checking, the qualified genomic DNA was used to construct a non-replication large-fragment genomic library with ~ 20 Kbp fragment sizes. The pooled library was bound to a polymerase and loaded onto a PacBio Sequel, after which PacBio Sequel SMRT Sequencing was performed. Hi-C libraries were prepared with independently extracted genomic DNA of the U. aurantiacoatra sample, and an Illumina sequencing library was constructed with a mean fragment size of ~ 350 bp for further Illumina Hiseq X10 sequencing. Hi-C and Illumina Hiseq sequencing data were used for genome-assisted assembly. For genomic DNA from samples of U. sp. (SC-4) and D. longissima (SC-9), independent Illumina Hiseq2500 sequencing was performed with 300- and 380-bp sequencing libraries, respectively, which was used for further de novo genome assembly.
Genome resources available in public databases
Genome information of Evernia prunastri (Meiser et al. 2017), Usnea florida (https://mycocosm.jgi.doe.gov/Usnflo1/Usnflo1.home.html), and Alectoria sarmentosa (Liu et al. 2019a) used for comparative genome analysis were download from JGI (Grigoriev et al. 2014) and NCBI database. Genome information of Letharia lupina (McKenzie et al. 2020), Imshaugia aleurites (PRJEB42325), Gomphillus americanus (PRJEB42325), Alectoria fallacina (PRJEB42325), Bacidia gigantensis (Allen et al. 2021), Physcia stellaris (Wilken et al. 2020), Letharia columbiana (McKenzie et al. 2020), and Lasallia pustulata (Merges et al. 2023) used for analyzing amino acid residues of positively selected genes were downloaded from NCBI database.
Genome assembly and annotation
The initial genome assembly was performed using FALCON software (Chin et al. 2013). For Hi-C data, Bowtie2 (v.2.3.2) (Langmead and Salzberg 2012) and HiC-Pro (v.2.8.1) (Servant et al. 2015) were applied for mapping reads, identifying valid interaction paired reads, and clustering scaffolds, respectively. Contigs with depths less than 50 × coverage were filtered out to exclude low-coverage genome sequences particularly potential algal genome sequences. FCS-GX was used to reconfirm the low degree of contamination from algae genomes (Astashyn et al. 2024). The final assembly was polished using independently sequenced Illumina Hiseq raw reads.
For protein-coding gene prediction, Augustus (Stanke et al. 2006) and GeneID (Parra et al. 2000) were used for de novo annotation, whereas GeMoMa (Keilwagen et al. 2016) and Genewise (Birney et al. 2004) were used for homologous annotation. The EVidenceModeler method (Haas et al. 2008) was used to integrate the two results for genomes sequenced in this study, including U. aurantiacoatra (NJ115-6), U. sp*.* (SC-4) and D. longissima (SC-9). The Funannotate (v1.8.13) with Usnea florida as the training model was used for genomes of Evernia prunastri and Alectoria sarmentosa. For D. longissima (SC-9), ORFs with both a start codon and a stop codon were retained to exclude incomplete annotations caused by short scaffolds.
For repeat sequence annotation, RepeatMasker (v.4.1.2) (Chen 2004), RepeatModeler (v.2.0.2) (Flynn et al. 2020), RepeatProteinMasker (v.4.1.2) (Chen 2004), and TandemRepeatFinder (v.4.09) (Benson 1999) were used with the repeat sequence database constructed with the U. aurantiacoatra (NJ115-6) assembly itself.
For ncRNA annotation, Rfam (Griffiths-Jones et al. 2005), tRNAscan-SE (Lowe and Eddy 1997), and RNAmmer (Lagesen et al. 2007) were used. For protein coding gene annotation, the first approach was to query the whole protein sequence to Swissprot (Bairoch et al. 2005) and KEGG (Ogata et al. 1999) databases, whereas the second approach was to use InterProScan (Zdobnov and Apweiler 2001) to identify conserved protein domains and annotate gene function. A circos plot (Krzywinski et al. 2009) was used to visualize the genomic landscape of related annotations.
Orthologous gene identifications by genome syntenic alignment
We performed genomic syntenic alignment for six lichen genomes, including U. aurantiacoatra (NJ115-6), U. sp. (SC-4), U. florida (ATCC18376), D. longissima (SC-9), E. prunastri (FR SP7-11), and A. sarmentosa (MAF-Lich 21536), using LASTZ (version 1.04.00) (Chen et al. 2019; Hecker and Hiller 2020). The genome of U. florida was used as the reference genome, and the other five genomes were aligned to it. The five pair-align results were combined to obtain a final multispecies alignment as a multiple alignment format file. Then, the corresponding U. florida annotated coding sequence was used as reference to retrieve orthologous DNA sequences from the multiple alignment format file. This dataset of orthologous sequences of the six species included 12,243 genes and was a start-up orthologue dataset for further analyses.
Evolutionary genetic analyses for adaptive selection
For positive selection analyses using PAML (v4.8), two tree-setting strategies were considered. Focusing on U. aurantiacoatra as the foreground branch, strategy #1 used all five species as background branches, and strategy #2 excluded D. longissima from the five species, using only four species as background branches. The reason to consider using strategy #2 was because the obvious fast growth rate of D. longissima (1–3 cm per year) compared with U. aurantiacoatra (4.3–5.5 mm per year) (Keon 2009; Esseen et al. 1981; Jansson et al. 2009; Li et al. 2014b). The results of the two tree strategies were combined to a final result. For each strategy, the same “more than 20% of the alignment in a gap” screening criterion was implemented. The result dataset of the two strategies included 5674 and 6202 orthologous genes, respectively, and their concatenated set was 6226 genes.
Based on the reconstructed phylogenomic tree, positively selected genes (PSGs) were identified using the branch-site model of the codon evolution with model = 2 and Nssites = 2, whereas for the branch model, the parameters were the null model (model = 0) versus the alternative model (model = 2). Genes with omega value (dN/dS) larger than 1 and statistically significant between foreground and background branch were regarded as PSGs. In PAML, a chi-square test is used to exam the statistical significance of the omega larger than 1. Besides, an FDR test was performed to exclude potential false positive PSGs.
To further confirm the target PSGs and find its potential amino acid residues where positive evolution occurs, eight additional lichen species with available genome data from Lecanoromycetes were added, including Letharia lupina (McKenzie et al. 2020), Imshaugia aleurites (PRJEB42325), Gomphillus americanus (PRJEB42325), Alectoria fallacina (PRJEB42325), Bacidia gigantensis (Allen et al. 2021), Physcia stellaris (Wilken et al. 2020), Letharia columbiana (McKenzie et al. 2020), and Lasallia pustulata (Merges et al. 2023). Orthologous genes of the target PSGs were identified from the total 14 genomes by a reciprocal BLAST strategy of INPARANOID algorithm (Remm et al. 2001). The potential amino acid residues were aligned to check their evolution and possible substitution variations.
Protein interaction network
Protein interaction network analyses were performed in EMBL’s STRING (http://string.embl.de). Four different fungal model species were implemented as background genomes for analyses, including Aspergillus nidulans, Cryptococcus neoformans, Neurospora crassa, and Saccharomyces cerevisiae. For analyses the PSGs in U. aurantiacoatra, a 0.40 default interaction score of medium confidence was used. Domain analyses were performed in the SMART online website (http://smart.embl.de/smart/), with PFAM and signal peptide items checked.
Generation of ΔUmrgs1 mutant for functional verification
The UaRGS1 ortholog in Umbilicaria muhlenbergii, UmRGS1, was identified by searching the genome (unpublished data) using a reciprocal BLAST strategy (Remm et al. 2001). The split-marker approach was applied to knock out UmRGS1 in U. muhlenbergii. Upstream and downstream flanking sequences of UmRGS1 were amplified using primers 1F/2R and 3F/4R (Table S2), respectively. The resulting PCR fragments were ligated onto the hph cassette amplified from pCX63 (Zhao et al. 2005) with primers HT-F/HY-R and YG-F/HT-R. The final UmRGS1 gene replacement fragments were transformed into protoplasts generated by Driselase (Sigma-Aldrich), as described previously (Wang et al. 2020). Hygromycin-resistant transformants were screened by using PCR with primers 5F/6R, 7F/HY-R, YG-F/8R, and H850/H852 (Fig. S1).
The lichen-forming fungus U. muhlenbergii strain (wild type) and UmRGS1 gene knockout mutant strains (LR2, LR5, and LR8) were routinely cultivated on potato dextrose agar medium at 20 °C for 7 days. To detect the survival rate of ultra-low-temperature stress, 2 × 10^3^ cells of the U. muhlenbergii JL3 strain and ΔUmrgs1 mutants were subjected to six freeze–thaw cycles of liquid nitrogen without any protective agent. Cold-shocked cells were recovered on PDA medium at 20 °C for 10 days. Each experiment was performed in triplicate to count the number of colonies on the recovered plates.
RESULTS
De novo genome assembly combined multi-sequencing technologies yield high-quality genome of Usnea aurantiacoatra
To obtain a high-quality reference genome, we sequenced Antarctica U. aurantiacoatra (NJ115-6) (Fig. 1a-1b) using three sequencing techniques of PacBio, HiSeq (Illumina) and Hi-C. First, we assembled the genome sequences with 61.01 Gbp PacBio data using FALCON, and obtained a draft genome with a size of 81.0 Mbp (187 contigs; contig N50 944.85 Kbp) after removing the potential contaminant and redundant sequences. Then, we corrected the contigs with 86.83 Gb HiSeq data using pilon (v.1.24) (Walker et al. 2014). Finally, we linked the corrected contigs to scaffolds with 73.71 Gb Hi-C data using Bowtie2 (v.2.3.2), HiC-Pro (v.2.8.1), and LACHESIS (Burton et al. 2013), and obtained a total of 81.4 Mb genome sequences (121 scaffolds; Scaffold N50 1.59 Mb; Fig. 1c; Fig. S2; Table 1).Table 1. Genome assembly statistics of usneoid lichens sequenced in this studyU. aurantiacoatra (NJ115-6)U. sp. (SC-4)D. longissimi (SC-9)Total Bases (Gbp)86.839.8311.50Assembly size (Mbp)81.4040.6067.30No. Scaffolds121896659,938Scaffolds N50 (Kbp)159013.901.80BUSCO (%)97.3688.5154.22Duplicated BUSCO (%)16.410.471.70Predicted genes13,53413,19112,475AccessionGWHBJEF00000000GSA: CRA007127GSA: CRA007127
We also performed Hiseq sequencing of the genome of two other usneoid lichen U. sp. (SC-4) and D. longissima (SC-9). We sequenced the genomic DNA (9.83 Gb for SC-4; 11.5 Gb for SC-9), and assembled a total of 40.6 Mb of SC-4 (contig N50 10.5 Kbp; scaffold N50 13.9 Kbp) and 67.3 Mbp of SC-9 (contig N50 1.3 Kbp; scaffold N50 1.8 Kbp) genome sequences using SOAPdenovo2, respectively (Table 1). Gene annotation obtained 13,534 protein-coding genes for U. aurantiacoatra, 13,191 genes for U. sp., and 12,475 genes for D. longissima (Table 1).
To carry out a comparative genome analysis, genome of Usnea florida (ATCC18376), Evernia prunastri (FR SP7-11), and Alectoria sarmentosa (MAF-Lich 21536), which belong to the same family (Parmeliaceae) as the three usneoid lichens mentioned above, were selected from the published genomic data. Of these six genomes, U. aurantiacoatra genome was the largest, almost double the size of the LFF genomes that have been published so far. To uncover the reason for the highly expanded genome size of U. aurantiacoatra, repeat sequence annotation was performed in these six LFF genomes. Comparison showed that the repeat sequences of U. aurantiacoatra were significantly higher than those of the other five genomes, with the greatest expansion of long terminal repeat (LTR) and unknown repeat sequences (Fig. 1d). The total size of all types of repeat sequence was about 40 Mbp, the same as the genome sizes of other lichen species except U. aurantiacoatra. Considering similar gene number in the six genomes (Table 1), it can be concluded that the large genome size in U. aurantiacoatra is due to amplification of repeat sequences. We also performed BUSCO assessment of the three usneoid LFF genome data obtained in this study, U. aurantiacoatra has the more complete genome (Fig. S3).
Genome signatures for adaptive selection of U. aurantiacoatra
In search of adaptive selection traits at genomic level, 6226 concatenated genes obtained by two tree-setting strategies (Fig. 2a) were used for positive selection analysis by PAML, and were corrected the p-values with false discovery rate (FDR) analysis (Fig. 2b). There were 178 PSGs with corrected p-values < 0.1 under both strategies (Fig. 2b), removing 35 pseudogenes yielded 143 PSGs (Fig. 2c), which included 86 PSGs paired with genes annotated by EVidenceModeler (Table S3).Fig. 2. Positive selection related to the adaption to extreme Antarctic environments. a Workflow of evolutionary genetic analyses for adaptive selection. Genomic syntenic alignment found 12,243 orthologs in six genomes, which were filtered for two tree-building strategies. The concatenation obtained by both strategies was used for adaptive analysis, resulting in 178 potential positively selected genes (PSGs). b The p-values of the potential PSGs obtained by two strategies were corrected for false discovery rate (FDR) analysis. Red and blue dots indicated PSGs with p-values < 0.1 in strategy I and II, respectively. Yellow dots were PSGs with corrected p-values < 0.1 under both strategies, which were used for subsequent analysis. c Pie chart showing the number of pseudogenes, annotated genes, and non-annotated genes in 178 potential PSGs. d Box plot of mean dN/dS shows a significantly high mean ω value in U. aurantiacoatra (p < 0.01)
We calculated ω ratios (dN/dS) for 6226 orthologous genes, and comparisons revealed that the average value of the ω ratios for U. aurantiacoatra was significantly higher than that of other LFF (p < 0.01, Fig. 2d, Table S4–S5), indicating that negative selection in this species is universally more relaxed than other species.
Functional enrichment and protein interaction network of positively selected genes
To acquire the biological processes and functions in which the above PSGs were involved, we performed functional enrichment analysis using clusterProfiler (Wu et al. 2021) with GO categories and KEGG pathways. By functional enrichment of 86 PSGs with a Q-value < 0.05, it showed that most of these proteins were associated with transmembrane transporter functions and vacuole components, and cyanoamino acid metabolism was also enriched (Table S6).
To identify potential functional information related to adaptive selection, protein-associated network analysis was carried out with EMBL’s STRING platform using four model fungal species (Aspergillus nidulans, Cryptococcus neoformans, Neurospora crassa, Saccharomyces cerevisiae). Of the 4 model fungi species in STRING, we identified 6 protein interaction networks, which included 35 genes of the 86 PSGs (Table S7). PSGs in four of these protein interaction networks were enriched in the GO category described above, which were related to transmembrane transporter activity or/and vacuole component (Fig. 3). The other two protein interaction networks that were not enriched by KEGG or GO were the G-protein-signaling and RNA helicase (Fig. 3). The RNA helicase network involves 12 proteins with functions related to the processing and maturation of rRNA and tRNA modification in the initiation of protein translation (Fig. 3). The G-protein-signaling network contains three proteins, regulator of G-protein-signaling, casein kinase I, and carboxypeptidase (Fig. 3).Fig. 3. Protein interaction networks of positively selected genes (PSGs) in Usnea aurantiacoatra. A total of 35 PSGs were in six protein interaction networks, four of which were enriched for vacuole component and transmembrane transporter activity. In addition to this, a larger protein interaction network centered on RNA helicase, and another network of three proteins centered on regulator of G-protein signaling
Effects of regulator of G-protein-signaling in cold resistance
To characterize PSGs as evidence of environmental adaption in Antarctic U*. aurantiacoatra*, we selected regulator of G-protein-signaling to further study, and named this protein as UaRgs1. Since U. aurantiacoatra cannot be genetically manipulated, we chose to knock out the homologous of UaRGS1 in U**mbilicaria muhlenbergii (UmRGS1). Three separate ΔUmrgs1 deletion mutants (LR2, LR5, and LR8) were generated and validated by PCR using anchor primers (Fig. S1; Table S2). We chose LR2 as the representative of mutants for the further cold resistant verification considering the consistent phenotype among the three mutants. Knockout of the UmRGS1 did not affect the growth phenotype of U. muhlenbergii. To determine whether UmRgs1 was critical for cold shock, the liquid nitrogen freeze–thaw experiment was performed on the wild-type strain and ΔUmrgs1 mutants. After three parallel experiments, the average number of wild-type and ΔUmrgs1 mutant colonies was 94 and 33 on the recovery medium, respectively (Fig. 4a; Table S8). The survival rate of wild-type strain (4.7%) was nearly three times that of the ΔUmrgs1 mutant (1.65%), with a significant difference between them (p = 0.001), indicating that the ΔUmrgs1 mutant is more sensitive to rapid freeze–thaw temperature changes than the wild-type. Therefore, we can deduce that UaRgs1 may have similar and even stronger function in Antarctic U. aurantiacoatra for resistance of the freeze–thaw sharp temperature changes.Fig. 4. Functional verification of UmRGS1 in rock tripe Umbilicaria muhlenbergii. a Survival colonies and survival rate of wild-type strain and ΔUmrgs1 mutant after six liquid nitrogen freeze–thaw cycles. Error bars represent SD, unpaired T-test with Welch’s correction, p = 0.001, n = 3 independent experiments. b Domain architecture of UaRgs1 and the locations of three amino acid residues undergo positive evolution. c Phylogenetic tree constructed by UaRgs1 with its homologous in 13 other lichen-forming fungal genomes. List on the right were the amino acids and corresponding codons for the three positive selection sites in UaRgs1. d Multiple alignment of the same set of protein sequences, with asterisks marking the sites of three amino acids, and amino acid position 519 was in conserved domain
We also identified potential amino acid residues where positive evolution occurs on UaRgs1 (Fig. 4b). A phylogenetic tree and multiple alignment using additional UaRgs1 orthologous in 13 LFFs, showed that amino acid 519 site, located in the DEP domain, was highly conserved but changed in Antarctic U. aurantiacoatra (Fig. 4c-4d), suggesting that the adaptive selection is associated with the biochemical and molecular function of the gene. Another branch site analysis for these 14 Rgs genes was conducted and the outcome showed that site 519 was under positive selection, which enhanced out conclusion.
DISCUSSION
The three poles, Antarctica, Arctic and the Third Pole (i.e., the Tibetan Plateau and its surroundings), are the coldest environment on the Earth, of which Antarctica is the most severe one and recorded the lowest air temperature. Lichens, as the pioneer organisms, can dominantly survive in such extreme environments and contribute substantial biodiversity of their flora, for which a set of cold tolerance mechanisms are needed. Previous studies on the Antarctic lichens mainly focused on the correlation of lichen biodiversity and conservation with climate change (Sancho et al. 2017; Romeike et al. 2002; Wauchope et al. 2019). A related study revealed differences in biosynthetic gene clusters between Umbilicaria pustulata lichens in Mediterranean and cold-temperate regions (Singh et al. 2021). This suggests that there must be a large number of environmental adaptation genes in Antarctic lichen genome. In this case, Usnea aurantiacoatra was chosen as a model mainly due to our field investigation and the good matching between divergent time of this lichen and the paleoclimate and paleogeology of Antarctica discovered by our study, which indicated that it is very likely to have a strong adaption to the Antarctic extreme environment driven by natural selection.
The approaches identifying natural selection at the molecular evolution level involves estimation of synonymous and nonsynonymous substitution rates and detection of positive selection in protein-coding DNA sequences among species (Yang 2007). We comprehensively compared the genomes between U. aurantiacoatra and those lichens mainly living in the temperate regions, then attempted to remove D. longissima with different growth rates when analyzing PSGs. The results of the two strategies were slightly different (Fig. S4), but the results obtained from different background branches may be both meaningful. We used a combination (the union) of the two strategies to ensure that genes selected by either strategy would be included in the final set of result genes.
The identified 86 PSGs possibly related to cold adaptation mainly enriched in lytic and storage vacuole components. The fungal vacuole serves as both the main compartment for division and a reservoir for the storage of small molecules, such as polyphosphate, amino acids, divalent calcium, and other small molecules (Klionsky et al. 1990). In some fungi, genes controlling the formation of vacuole play an important role in resistance to stresses (Son et al. 2018; Wilken et al. 2020; Zhu et al. 2023). Antarctic lichens need to undergo long periods of dormancy, and it is hypothesized from our results that U. aurantiacoatra enhance its vacuole function under selective pressure.
Two other potentially important mechanisms were found through protein network analysis. RNA helicase at the center of the largest protein interaction network, has been intensively studied in both eukaryotes (O’Day et al. 1996) and bacteria (Liu et al. 2021). This gene involves in the maturation of 35S-pre-rRNA and is required for cleavages leading to mature 18S rRNA in cell nucleus, which plays a critical role for the biogenesis of ribosomes. RNA helicase has been shown to be related to thermo-tolerance in rice (Wang et al. 2016) and cold tolerance in glacial psychrophilic Flavobacterium (Liu et al. 2021). Besides, the tRNA methyltransferase gene interacted with RNA helicase (Fig. 3) has also been verified to be related with temperature changes in bacteria and yeast (Lorenz et al. 2017, Hori 2014). In LFF Cladonia grayi, rRNA and the biogenesis of ribosomes have been suggested to be closely related with stress resistance (Armaleo et al. 2019). Our results provide further evidence of the importance of the RNA helicase associated protein network in resistance.
In this study, we chose to perform an extended analysis of regulator of the G-protein signaling (RGS) in another protein network, because G-protein and RGS belong to important pathways that receive external signals in fungi (Lengeler et al. 2000; Zhong and Neubig 2001; McCudden et al. 2005). Orthologous genes of UaRGS1 involves in the regulation of growth, sporulation, and pathogenicity in model fungi (Chan and Otte 1982; Yu et al. 1996; Ballon et al. 2006; Liu et al. 2007), but there is no experimental evidence that it is related to external low-temperature stimuli or other environmental stress. Here, we verified the new function of RGS related to the ultra-low temperature tolerance. Furthermore, it is worth mentioning that there is a casein kinase I gene in the UaRgs1 protein interaction network (Fig. 3). Casein kinase I is conserved from plants to animals, it performed a number of cellular processes, including DNA repair, cell cycle, cytokinesis, vesicular trafficking, and circadian rhythm (Park et al. 2012; Marzoll et al. 2022). The protein interaction network analysis allows further enrichment of PSGs and can shed more light on the cellular processes in which environmentally adapted genes are involved. The identification of RGS and casein kinase I interaction network in PSGs is more indicative of G-protein signaling pathways that were subjected to selective pressures. Both RGS and casein kinase I are important factors upstream of cellular signaling pathways, and in the case of RGS, as a negative regulator of G-proteins, it is responsible for making the cell to respond correctly to a variety of signals, not just adaptation to cold stress, and it is possible that it will have similar results across different external stresses.
Besides, there is also some other genomic signatures suggesting that the LFF genome is under adaptive selection. The assembly size of U. aurantiacoatra is two times as large as those of other relative lichen species, which is caused by LTR amplification and unknown repeat sequences. The LTR amplification is one of the main drivers leading to the big size of eukaryotic genome (Liu et al. 2019b). LTRs show considerable enrichment in lncRNA transcripts compared with non-LTR elements in mouse and human (Kannan et al. 2015; Kapusta et al. 2013; Kelley and Rinn 2012). Most of LTRs transcribed in lncRNAs serve as exons (Kannan et al. 2015), specific families have been co-opted as promoters (Thompson et al. 2016). In plants such as mangrove trees (Lyu et al. 2018), palm (Schley et al. 2022) and bamboo (Papolu et al. 2021), LTR copy number has been reported to crucially associated with adaptation to various environmental stress including aridity or heat. Plenty of evidence suggests that LTR in Plants involves in the modulation of gene expression in response to several stimuli, notably stresses and external challenges (Bui and Grandbastien 2012). In Medicago sativa, LTR has been reported to be related to cold tolerance. Study of Sicilian blood oranges (Citrus sinensis) illustrated the strength of the LTR as a promoter and as an upstream activating sequence is of cold dependency (Butelli et al. 2012). Therefore, it is reasonable to presume that in the U. aurantiacoatra as the symbiont of lichen-forming fungus and plant (alga), the existence of abundant LTR is related to the tolerance of cold environment.
CONCLUSIONS
In summary, based on our genomic sequencing and bioinformatic analyses, we exhibited several potential mechanisms on how U. aurantiacoatra achieved its adaptation to Antarctic environments. We also performed functional validation of one of the genes that has not yet been reported to be associated with stress, further determining the accuracy of the screened PSGs. This study can serve as a good start to understand the environmental adaptions of lichens, and provide the basis and clues for further studies on LFF resistance mechanisms.
Supplementary Information
Supplementary Material 1: Figure S1. Gene replacement of UmRGS1. a Diagram of the UmRGS1 gene and primers used to generated the ΔUmrgs1 mutant. HY and YG are fragments of the hph cassette conferring resistance to hygromycin. b Mutant detected with four pairs of anchor primers. Figure S2. GC content-sequencing depth distribution of three usneoid lichen-forming fungi. a The left and right panels represent the results of U. aurantiacoatra (NJ115-6) using Illumina and Nanopore sequencing platforms, respectively. b GC-depth of Illumina-sequencing U. sp. (SC-4). c GC-depth of Illumina-sequencing D. longissima (SC-9). Figure S3. Integrity and characterization of U. aurantiacoatra genome. a BUSCO assessment of three usneoid LFF genomes with the highest completeness of U. aurantiacoatra. b Gene feature distribution of U. aurantiacoatra. Figure S4. The differences between ω and p values of two strategies. a Comparison of ω values of two strategies. b Comparison of p values of two strategies.Supplementary Material 1: Table S1. Information on lichen samples used in this study. Table S2. PCR primers used in this study. Table S3. List of positively selected genes and annotation in Antarctic Usnea aurantiacoatra. Table S4. Results of branch site analysis for strategy 1. Table S5. Results of branch site analysis for strategy 2. Table S6. Results of positively selected genes enrichment in GO ontology and KEGG pathway in Antarctic Usnea aurantiacoatra. Table S7. Protein interaction networks and annotation of positively selected genes in Antarctic Usnea aurantiacoatra. Table S8. Survival colonies and survival rates of WT and the ΔUmrgs1 mutant after ultra-low-temperature shock.
The reference list from the paper itself. Each links out to its DOI / PubMed record.