Gut bacterial community profile of frugivorous bats from Cathedral Cave in Cavinti, Laguna, Philippines
Bonie B. Datul, Ronilo Jose D. Flores, Andrew D. Montecillo, Marian P. De Leon

TL;DR
This study analyzes the gut bacteria of fruit-eating bats in a Philippine cave, finding Proteobacteria and Firmicutes as the most common.
Contribution
The paper provides a novel characterization of gut microbial communities in frugivorous bats from a specific Philippine cave.
Findings
Proteobacteria and Firmicutes were the most abundant phyla in the bats' gut microbiota.
The study highlights the microbial diversity associated with frugivorous bats in a cave ecosystem.
Abstract
Here we report the 16S rRNA gene amplicon analysis of the gut microbiota of three frugivorous cave bat species from the Cathedral Cave in Cavinti, Laguna, Philippines. Among the bat species, the most abundant phyla are Proteobacteria and Firmicutes D.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
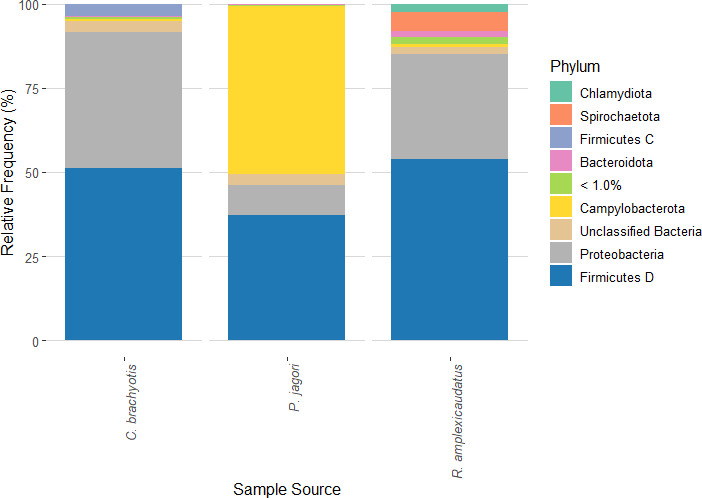 Fig 1
Fig 1| Library name | Host bat species | Input | Filtered | Denoised | Merged | Input merged | Non- chimeric | Percentage of bacterial sequences (%) | SRA accession no. |
|---|---|---|---|---|---|---|---|---|---|
| Cbra1001 |
| 109,480 | 91,267 | 91,005 | 90,085 | 82.28 | 75,820 | 69.25 |
|
| Cbra1002 |
| 119,356 | 102,065 | 101,940 | 101,877 | 85.36 | 101,525 | 85.06 |
|
| Cbra1003 |
| 103,575 | 86,831 | 86,665 | 86,265 | 83.29 | 82,051 | 79.22 |
|
| Cbra1004 |
| 130,024 | 110,188 | 110,031 | 109,589 | 84.28 | 106,043 | 81.56 |
|
| Pjag1001 |
| 107,007 | 86,977 | 86,630 | 86,425 | 80.77 | 84,759 | 79.21 |
|
| Pjag1002 |
| 60,987 | 45,914 | 45,488 | 44,751 | 73.38 | 43,271 | 70.95 |
|
| Pjag1003 |
| 164,588 | 142,256 | 142,195 | 142,137 | 86.36 | 141,670 | 86.08 |
|
| Pjag1004 |
| 119,773 | 103,600 | 103,313 | 102,229 | 85.35 | 97,581 | 81.47 |
|
| Pjag1005 |
| 107,308 | 91,385 | 91,329 | 91,154 | 84.95 | 90,336 | 84.18 |
|
| Ramp1001 |
| 131,413 | 107,852 | 107,593 | 106,988 | 81.41 | 101,948 | 77.58 |
|
| Ramp1002 |
| 117,861 | 97,092 | 96,924 | 96,089 | 81.53 | 92,275 | 78.29 |
|
| Ramp1003 |
| 129,725 | 107,828 | 107,726 | 107,628 | 82.97 | 107,057 | 82.53 |
|
- —Department of Science and Technology, Philippines (DOST)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBat Biology and Ecology Studies · Human-Animal Interaction Studies · Gut microbiota and health
ANNOUNCEMENT
Bats inhabit diverse ecological niches worldwide, from thick rainforests to busy urban areas. Despite their ecological prominence, their gut microbiota composition remains largely unexplored. The gut plays a pivotal role in bat health, metabolism, and immune function, and unraveling the composition and dynamics of the bat gut microbiota has important implications for understanding host-microbe interactions and their roles on bat physiology and ecology (1, 2). Here, we present the gut-associated bacterial community of three frugivorous bat species, namely, Cynopterus brachyotis (Müller, 1838), Ptenochirus jagori (Peters, 1861), and Rousettus amplexicaudatus (E. Geoffroy, 1810) from the Cathedral Cave, Cavinti Underground River and Caves Complex, Cavinti, Laguna, Philippines (14°16′54.48″ N, 121°38′9.661″ E) through 16S rRNA metabarcoding.
The gut of three bat species: C. brachyotis (n = 4), P. jagori (n = 5), and R. amplexicaudatus (n = 3) were collected in November 2021. The gut was removed at the sampling site then transported to the laboratory in liquid nitrogen. The intestines were opened, and the mucosal lining tissue with the gut contents was scraped off (3, 4) for DNA extraction using Microbiome DNA Isolation Kit (Norgen Biotek, ON, Canada). The lysis step was modified to 10 minutes at 65°C. Libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA) following the manufacturer’s 16S Library Preparation Workflow: the V3-V4 regions of the16S rRNA gene were amplified using 341F and 805R primers with Illumina adapters (5). Sequencing was performed on Illumina MiSeq (2 × 300 bp) (5).
The demultiplexed sequence data were processed using QIIME2 (v. 2023.9) (6). Default parameters were used unless otherwise stated. DADA2 (v. 2023.9.0) (7) was used to trim the reads, resolve amplicon sequence variants (ASVs), and merge overlapping sequences. Reads were truncated based on the values obtained from FIGARO (8): p-trunc-len-f and p-trunc-len-r for C. brachyotis, P. jagori, and R. amplexicaudatus are 284 and 239, 289 and 234, and 296 and 227, respectively; p-max-ee-f and p-max-ee-r for all samples are both 2. Forward and reverse reads were trimmed at positions 17 and 21, respectively. Taxonomy was assigned using the multinomial naive Bayes classifier method (9) in the q2-feature-classifier plugin (v. 2023.9.0) (10) using the V3-V4 region from the Greengenes2 (v. 2022.10) reference database (11, 12). Non-bacterial ASVs were removed using the q2-taxa (6). Resulting ASVs were combined per bat species with the merge function in qiime2 (6) and used for downstream analyses in R (v.4.3.3) (13) using the phyloseq (14) and ggplot2 (15) packages.
A combined total of 462,435, 559,663, and 378,999 paired-end reads were obtained from C. brachyotis, P. jagori, and R. amplexicaudatus, respectively (Table 1). The three most abundant phyla are Firmicutes D (51.01%), Proteobacteria (40.47%), and Firmicutes C (3.52%) for C. brachyotis, Campylobacterota (50.08%), Firmicutes D (37.07%), and Proteobacteria (8.99%) for P. jagori, and Firmicutes D (53.74%), Proteobacteria (31.32%), and Spirochaetota (5.38%) for R. amplexicaudatus (Fig. 1). It is noteworthy that the gut bacterial community diversity among the bat species was different.
Taxa bar plots showing representative phyla composing the gut-associated bacteria of the three fruit bat species collected from Cavinti Underground River and Caves Complex determined by amplicon sequencing of the V3-V4 region of the 16S rRNA. Each bar represents the combined data from specific bat species, and each colored stacked box represents a bacterial taxon.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Carrillo-Araujo M, Taş N, Alcántara-Hernández RJ, Gaona O, Schondube JE, Medellín RA, Jansson JK, Falcón LI. 2015. Phyllostomid bat microbiome composition is associated to host phylogeny and feeding strategies. Front Microbiol 6:447. doi:10.3389/fmicb.2015.0044726042099 PMC 4437186 · doi ↗ · pubmed ↗
- 2Li J, Li L, Jiang H, Yuan L, Zhang L, Ma J-E, Zhang X, Cheng M, Chen J. 2018. Fecal bacteriome and mycobiome in bats with diverse diets in South China. Curr Microbiol 75:1352–1361. doi:10.1007/s 00284-018-1530-029922970 · doi ↗ · pubmed ↗
- 3Nordgård L, Traavik T, Nielsen KM. 2005. Nucleic acid isolation from ecological samples--vertebrate gut flora. Methods Enzymol 395:38–48. doi:10.1016/S 0076-6879(05)95003-915865959 · doi ↗ · pubmed ↗
- 4Phillips CD, Phelan G, Dowd SE, Mc Donough MM, Ferguson AW, Delton Hanson J, Siles L, Ordóñez-Garza N, San Francisco M, Baker RJ. 2012. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol Ecol 21:2617–2627. doi:10.1111/j.1365-294X.2012.05568.x 22519571 · doi ↗ · pubmed ↗
- 5Illumina. 2013. 16S metagenomic sequencing library preparation
- 6Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al.. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi:10.1038/s 41587-019-0209-931341288 PMC 7015180 · doi ↗ · pubmed ↗
- 7Callahan BJ, Mc Murdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA 2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi:10.1038/nmeth.386927214047 PMC 4927377 · doi ↗ · pubmed ↗
- 8Weinstein MM, Prem A, Jin M, Tang S, Bhasin JM. 2020. FIGARO: an efficient and objective tool for optimizing microbiome r RNA gene trimming parameters. Bioinformatics. doi:10.1101/610394 · doi ↗