Thermodynamic Origin of the Linear Pressure Dependence of DNA Thermal Stability
Jurij Lah, San Hadži

TL;DR
This paper explains why DNA's thermal stability changes linearly with pressure, linking it to DNA hydration and thermodynamic properties.
Contribution
The study reveals the thermodynamic origin of DNA's linear pressure dependence through compressibility and expansibility analysis.
Findings
The linear pressure dependence of DNA stability is due to hydration effects on compressibility and expansibility.
A convergence temperature exists where compressibility and volume of (un)folding become zero.
Predictions show how temperature and pressure affect DNA's thermodynamic stability parameters.
Abstract
High pressure affects the structure and function of DNA and is a key parameter for studying the origin and physical limits of life. Different types of DNA structures systematically show a linear pressure dependence of thermal stability (up to ∼200 MPa), which is maintained even when the solution composition is changed. The reasons behind the linear pressure dependence are not understood. We have performed a thermodynamic analysis of the pressure-, temperature- and composition-dependent (un)folding of various polynucleotide duplexes and G-quadruplexes. We demonstrate that the reason for the observed linearity is the link between compressibility and expansibility, both of which largely depend on DNA hydration. We predicted the temperature and pressure dependence of compressibility and expansibility of (un)folding and explain how they affect the corresponding volume change and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
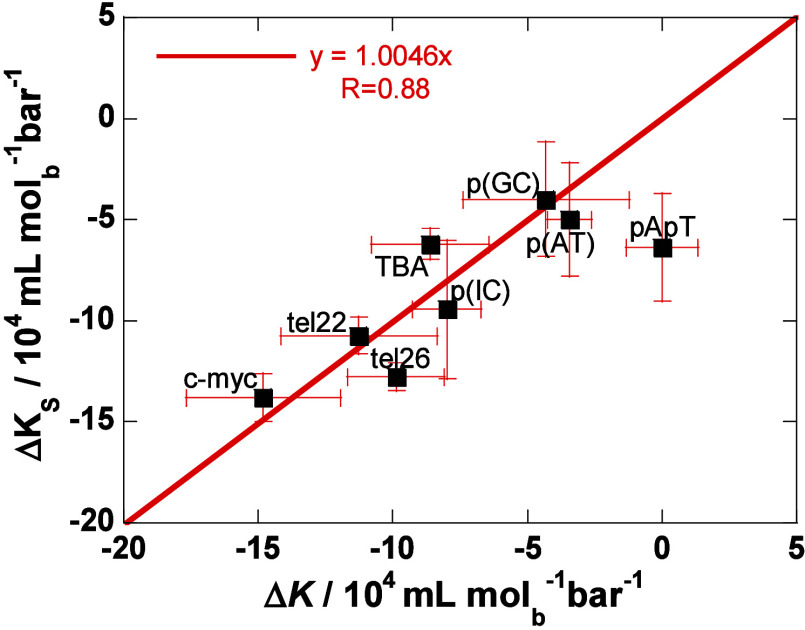 Figure 1
Figure 1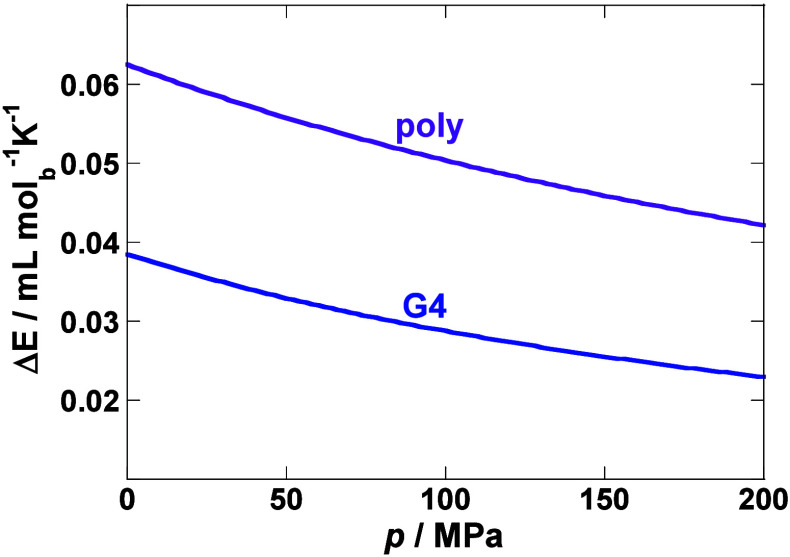 Figure 2
Figure 2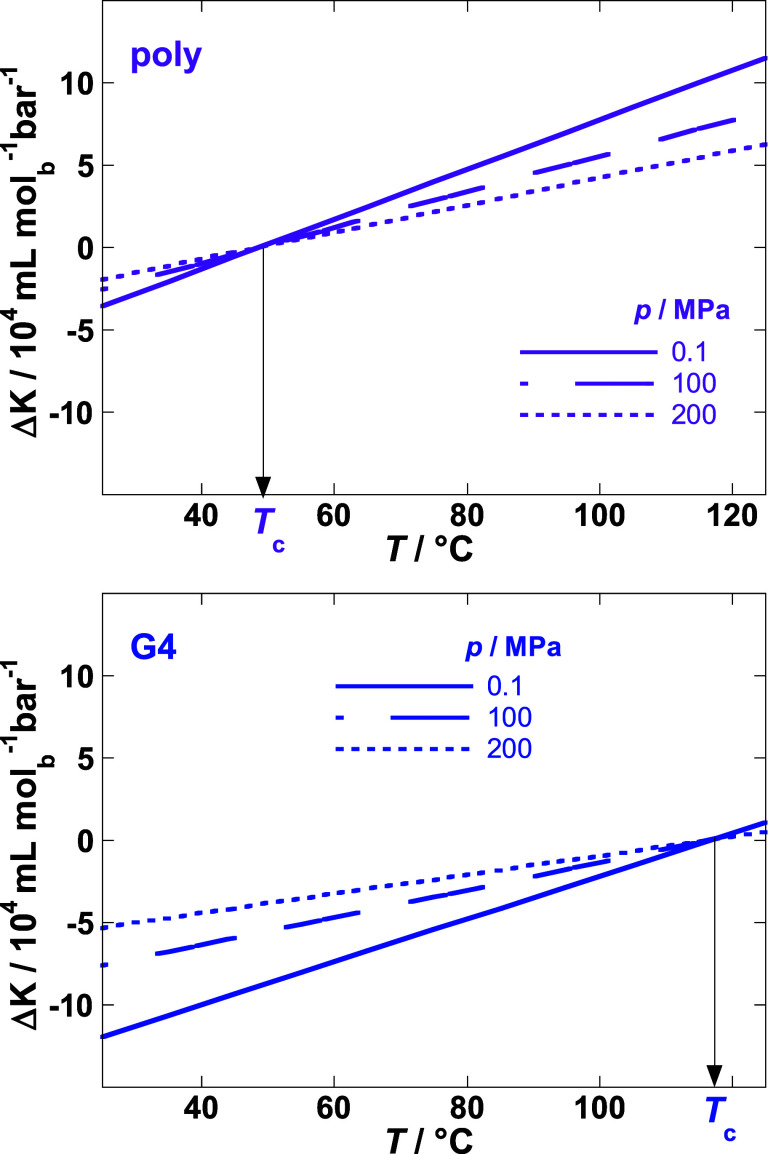 Figure 3
Figure 3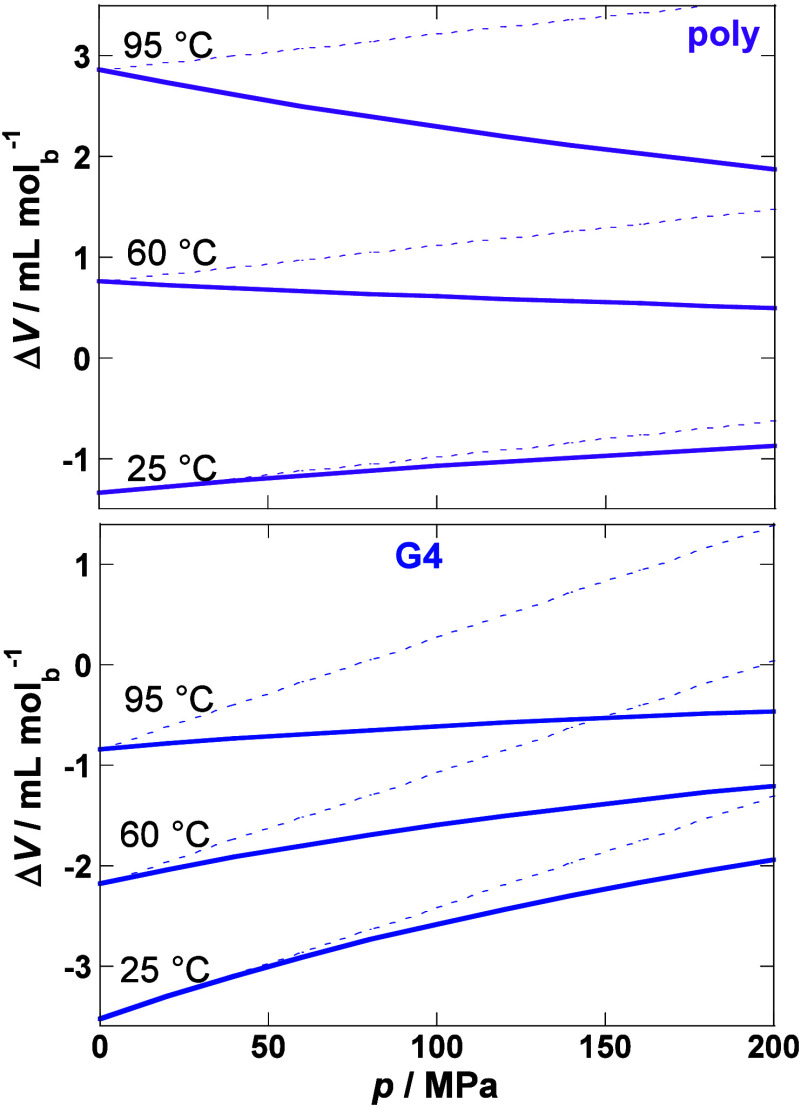 Figure 4
Figure 4 Figure 5
Figure 5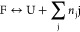 Figure 6
Figure 6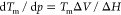 Figure 7
Figure 7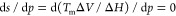 Figure 8
Figure 8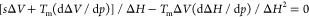 Figure 9
Figure 9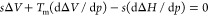 Figure 10
Figure 10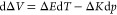 Figure 11
Figure 11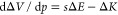 Figure 12
Figure 12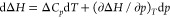 Figure 13
Figure 13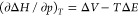 Figure 14
Figure 14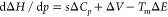 Figure 15
Figure 15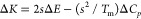 Figure 16
Figure 16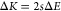 Figure 17
Figure 17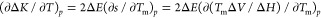 Figure 18
Figure 18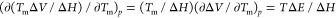 Figure 19
Figure 19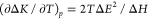 Figure 20
Figure 20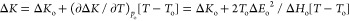 Figure 21
Figure 21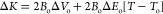 Figure 22
Figure 22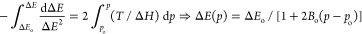 Figure 23
Figure 23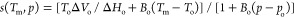 Figure 24
Figure 24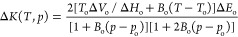 Figure 25
Figure 25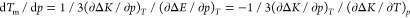 Figure 26
Figure 26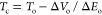 Figure 27
Figure 27- —Javna Agencija za Raziskovalno Dejavnost RS10.13039/501100004329
- —Javna Agencija za Raziskovalno Dejavnost RS10.13039/501100004329
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDNA and Nucleic Acid Chemistry · Protein Structure and Dynamics · Bacteriophages and microbial interactions
In native state biomacromolecules adopt unique three-dimensional structures that are required to perform their biological function under the normal life conditions. These three-dimensional structures are stabilized by relatively weak noncovalent intra- or intermolecular interactions that can be perturbed by changes in temperature, T, pressure, p, and the composition of the surrounding medium. The application of high pressures is of general physicochemical and biological interest, e.g. for understanding the physiology of organisms living in the deep sea.^1−9^ An increase in pressure changes the stability of protein and nucleic acid structures by shifting their population toward structures that occupy smaller volumes (Le Chatelier’s principle).^10−17^ The volumetric properties of biomacromolecules thus determine the effect of pressure on the equilibrium between the folded (F) and unfolded (U) states. For many biomolecules this equilibrium can be described by a two-state mechanism
which assumes that species j (e.g., ions, water molecules, cosolutes, cosolvents) exchange upon unfolding. Their apparent number, nj, may be positive (release of j) or negative (uptake of j). An increase in pressure shifts the equilibrium (eq 1) and consequently the thermal stability of the biomacromolecule (“melting” temperature), Tm, which is defined as the temperature at which the populations of F and U species are equal. At a constant concentration of species j, the pressure dependence of Tm can be described by the Clapeyron equation
where ΔV and ΔH represent the volume and enthalpy of unfolding, both of which depend on T and p. Therefore, dTm/dp is expected to vary with T and p, meaning that in general Tm versus p (pseudo)phase transition curves are not linear.^18−21^ However, for many different DNA structures a linear dependence of Tm on p is systematically observed in a broad range of pressures from 0.1 up to 200 MPa.^22−32^ In other words, why the slope, dTm/dp, is independent of p in this wide pressure range is not understood.
Here we examine the underlying thermodynamic reasons for this behavior. We demonstrate that the linear dependence of Tm on pressure (p) primarily arises from the specific relationship between compressibility and expansibility, both of which largely depend on DNA hydration.^33,34^ The presented relationship allows estimation of the compressibility of DNA unfolding using ΔV, ΔH and the corresponding expansibility, and suggests how compressibility and expansibility of unfolding depend on T and p. We also predict the existence of a convergence temperature at which ΔV, dTm/dp, and compressibility of unfolding simultaneously become equal to zero.
First, our analysis will provide thermodynamic reasons why the slope, dTm/dp, is independent of p at any selected solution composition. Hereafter we refer to the slope as s = dTm/dp. Pressure independence of s means that at constant solution composition:
Taking the derivative of eq 3 it follows that
Under the studied conditions the enthalpy of unfolding, ΔH, is always a positive quantity. Multiplication with ΔH leads to the expression:
The dependence of ΔV on T and p can be expressed as
where ΔE = (∂ΔV/∂T)p represents the expansibility of unfolding while ΔK = −(∂ΔV/∂p)T is the corresponding isothermal compressibility. At T = Tm dividing eq 6 by dp gives
The dependence of ΔH on T and p is given as
where is the heat capacity of unfolding. Moreover, the thermodynamic equation of state defines (∂ΔH/∂p)T as
Inserting eq 9 into eq 8 and dividing so obtained eq 8 by dp defines the pressure dependence of ΔH on the Tm versus p line as
The combination of eqs 5, 7 and 10 results in the relation
Equation 11 shows that the observed linear dependence of Tm on p postulates the relationship between ΔK, ΔE and ΔCp. Is the relation between ΔK, ΔE and ΔCp surprising? No, since for DNA unfolding all three quantities are closely related to changes in hydration. For duplexes and quadruplexes around room temperature ΔK < 0, ΔE > 0 and ΔCp > 0.^27,33^ The observed decrease in compressibility and increase in expansibility and heat capacity upon unfolding are in mutual agreement and suggest that the number of water molecules solvating the unfolded state is higher than that solvating the folded state, which is consistent with larger solvent accessible surface area of the unfolded state.^33^ For aqueous solutions, the isothermal compressibility, ΔK, is equal within experimental error to the corresponding adiabatic compressibility, ΔKS, which can be obtained from ultrasound velocity measurements.^33^ Experimental values for all these quantities (ΔK = ΔKS, ΔE, ΔCp, s) are known only for some polynucleotide duplex and G-quadruplex DNA.^22−27,34−40^ These data (presented in Table S1) suggest that in eq 11 the first term, 2sΔE, is the one that predominantly determines ΔK, while the second term, (s^2^/Tm)ΔCp, is significantly smaller in magnitude. By neglecting the second term the eq 11 simplifies into
In the following, we show that this equation enables a quantitative analysis of the dependence of ΔK and ΔE on temperature and pressure. Since the slope, s, is constant on the Tm versus p line, eq 12 suggests that ΔE and ΔK must have a specific dependence on pressure and temperature, which is investigated next. Ideal species behavior can be assumed for the dilute solutions used in (un)folding studies. Therefore, the partial molar quantities and the corresponding quantities of unfolding can be considered composition-independent. If Tm is varied by changing the composition of the solution at a given p (changing the concentration of species j, e.g. the concentration of sodium ions), then the corresponding ΔF change is only due to its temperature dependence and not due to the changed solution composition. This property can be used to evaluate the temperature dependence of ΔE. It has been observed that at p = const. the dependence of ΔV on Tm is linear,^22−31^ which means that its slope, ΔE, can be considered independent of T and the solution composition. Based on this the dependence of ΔK on T can be analyzed by the temperature derivative of eq 12:
Given that heat capacity of the unfolding ΔCp is a positive quantity, ΔH increases with increasing Tm. Thus, when the numerator Tm in the Tm/ΔH ratio increases, the denominator also increases to an extent that in the studied temperature interval the Tm/ΔH ratio can be considered constant within the experimental error. Under these assumptions, the derivative in eq 13 can be expressed as
while (∂ΔK/∂T)p as
Since T/ΔH and ΔE can be regarded as temperature-independent (see above), the derivative (∂ΔK/∂T)p can also be taken as a temperature-independent quantity. Thus, the integration of eq 15 (for p = p0) results in the relation
where ΔH0 = ΔH(T0,p0), ΔE0 = ΔE(p0) and ΔK0 = ΔK(T0,p0) are the quantities defined at the selected reference state (T0,p0). By expressing ΔK0 form eq 12, we obtain
where B0 = T0_ΔE0/ΔH0. Equation 17 thus predicts that the dependence of ΔK on T is linear, in agreement with the experimental observations.^34,38^ More importantly, eq 17 enables comparison between the calculated ΔK(T,p0) values with the corresponding experimental ΔKS(T,p0) values. In other words, ΔK(T,p0) can in principle be estimated from the corresponding ΔV, ΔEand ΔH values. To compare the validity of the estimated values at the same temperature, the experimental ΔKS_ measured at temperature TS (Table S1) were extrapolated to the selected T using eq 16: ΔKS = ΔKS(TS) + (∂ΔK/∂T)p0(T – TS). We observe a good agreement between experimental ΔKS and ΔK calculated using eq 17 (Figure 1), with the slope of the correlation line around one, which justifies the approximations applied in deriving eq 17. The largest deviation from the correlation line is observed for poly[d(A)]poly[d(T)] (Figure 1). For G-quadruplexes the predicted ΔK [given per mole of bases, 25 °C, 1 bar values between −6 × 10^4^ and −15 × 10^4^ mL mol_b_^–1^ bar^–1^] are significantly different from those of the polynucleotide duplexes [values between 0 and −10 × 10^4^ mL mol_b_^–1^ bar^–1^]. On the other hand, the slope (∂ΔK/∂T)p0 = appears to be positive and similar for the two sets of different DNA structures analyzed here [average value: (0.14 ± 0.06) × 10^4^ mL mol_b_^–1^ bar^–1^ K^–1^], which is in agreement with the previous observation that the temperature dependence of ΔK is relatively insensitive to the type of the studied DNA.^34^ Taken together, these findings suggest that the temperature dependence of ΔK can be successfully estimated using eq 17.
The next question is how ΔE depends on pressure and how this affects the temperature dependence of ΔK. Experimental observations suggest that ΔE can be considered as a positive, temperature-independent quantity at constant pressure. From Maxwell’s relation it follows that (∂ΔE/∂p)T = −(∂ΔK/∂T)p = −2TΔE^2^/ΔH. Accordingly, the dependence of ΔE on p at given T can be estimated by integration of the differential equation
where T/ΔH ratio was assumed to be pressure-independent (T/ΔH = T0/ΔH0). Figure 2 shows that ΔE decreases nonlinearly with increasing pressure. It should be noted that the inclusion of meaningful experimentally based estimates of the T/ΔH pressure dependence does not significantly change the predicted pressure dependence of ΔE.
Maxwell’s relation (∂ΔE/∂p)T = −(∂ΔK/∂T)p shows how the pressure dependence of ΔE affects the temperature dependence of ΔK. Using eq 12, ΔK(p) = 2s(p)ΔE(p), we will now analyze how pressure affects ΔK at constant temperature. Since we consider ΔK to be independent of the composition, the problem can be translated into analyzing ΔK as a function p at constant Tm. The fact is that a change in p is accompanied by a change in Tm at constant solution composition. Therefore, a change in p at constant Tm is only possible when the change in Tm due to a change in pressure exactly compensates the change in Tm due to change in composition. In this way, the equation for s(Tm, p) was derived (see SI)
The combination of eqs 12, 18, and 19 results in
where B0 = T_0_ΔE0/ΔH0. Equation 20 describes ΔK as a function of the independent variables T and p, which is for polynucleotide duplexes and G-quadruplexes presented in Figure 3.
Finally, we show that the ΔK versus T and p data (Figure 3) contain all the necessary information on the thermal stability of DNA as a function of pressure. Namely, the ratio of the derivatives −(∂ΔK/∂p)T/(∂ΔK/∂T)p, which can be estimated from these data, is directly related to dTm/dp (see derivation in SI)
Figure 3 shows that (∂ΔK/∂T)p is positive and decreases with increasing p. This is because ΔE decreases as p increases [(∂ΔK/∂T)p = 2TΔE^2^/ΔH, Figure 2]. On the other hand, (∂ΔK/∂p)T changes with T from positive values at T < Tc (⇒ dTm/dp < 0) to negative values at T > Tc (⇒ dTm/dp > 0), where Tc represents the convergence temperature at which ΔK reaches exactly zero. It follows from eq 20 that the convergence temperature Tc is independent of pressure and can be estimated as
Since ΔE > 0, it follows from eq 12 that ΔK = 0 when ΔV = 0 (resulting in s = 0). Thus, the observed linear dependence of Tm on p through the derived eq 12 postulates the existence of the convergence temperature at which ΔK, ΔV and s = dTm/dp simultaneously become equal to zero. The estimated Tc values for polynucleotide duplexes range between 45 to 65 °C (with the exception of poly[d(A)]poly[d(T)]), while for quadruplexes they range between 100 and 145 °C, which is in line with the reported temperature dependencies of ΔV and ΔK.^27,34,38^ Obviously, the mutual compensation of the contributions within ΔV and ΔK and their specific temperature dependencies cause ΔV and ΔK to change with temperature from negative (at T < Tc) to positive (at T > Tc) values. The prediction that this occurs at the same temperature (Tc) for both ΔV and ΔK suggests that hydration contributions play the most important role in this compensation.^29^ What causes the sign change of ΔV for DNA duplex unfolding at around 50 °C has been explained by Makhatadze, Marky and co-workers in the recent article.^41^ Following their analysis, ΔV can be considered as the sum of two contributions: ΔV= ΔVhyd + ΔVother. ΔVhyd represents the volume change associated with the interactions of the folded and unfolded states with water (i.e., hydration volume). ΔVother represents the contributions associated with the conformational changes of DNA (i.e., intrinsic volume change of the DNA + thermal volume change indicating the change of the void space). ΔVother appears to be temperature independent and has a negative sign. However, the ΔVhyd contribution is positive and increases with increasing temperature since expansibility ΔE, which is determined predominately by the hydration contribution,^33^ is a positive quantity. At T < Tc (∼50 °C) it appears that the positive values of ΔVhyd are not sufficient to overcome the negative values of ΔVother, so that ΔV < 0. Increasing the temperature increases ΔVhyd that overcomes the negative ΔVother at T > Tc. As a result, the value of ΔV becomes positive at temperatures above 50 °C. Why are the convergence temperatures for ΔV and ΔK very similar (equal within our approximations)? This is a direct consequence of the observed linearity which postulates that at a given pressure ΔK is proportional to ΔV (eq 12). ΔK itself is dominated by the hydration contribution^33^ that is negative at T < Tc and increases with temperature resulting in positive values of ΔK at T > Tc.
Why is Tc for G-quadruplexes very different (>50 °C higher) than for duplexes? In contrast to duplex, ΔVother for G-quadruplex unfolding has a positive sign.^27,35^ Its magnitude is significantly lower than that of ΔVhyd that has a negative sign,^27,35^ resulting in ΔV < 0 at lower temperatures. However, ΔVhyd becomes less negative at higher temperatures since expansibility ΔE, which is for G-quadruplexes also determined predominately by the hydration contribution,^27^ is a positive quantity. It appears that the reason that Tc (G-quadruplex) ≫ Tc (duplex) is 2-fold. At low temperature the compensation of the contributions results in ΔV that is significantly more negative for G-quadruplexes than for duplexes (Table S2). In addition, the expansibility that determines the temperature dependence of ΔV (and ΔK - eq 12) is significantly lower for G-quadruplexes (Table S2) meaning that increasing of ΔV and ΔK with temperature is less intense for G-quadruplexes than for duplexes. Interestingly, Tc for G-quadruplexes is very similar to the convergence temperature at which the entropy of transfer of hydrophobic groups of globular proteins from their core to water reaches zero.^42^ In other words, the volumetric properties of the unfolding of G-quadruplexes, which have a globular structure stabilized by dehydration, are more similar to those of globular proteins than to those of DNA duplexes.^43^
Taken together, our analysis suggests that the pressure and temperature dependencies of ΔK and ΔE need to be considered when extrapolating the thermodynamic parameters of DNA unfolding to different p and T. Figure 4 shows that they must be taken into account when extrapolating ΔV (see eq S16). In contrast, when extrapolating the enthalpy, ΔH, the entropy, ΔS, and the Gibbs free energy, ΔG, the consideration of the p and T dependence of ΔK and ΔE does not play a major role (Figure S2, eqs S17–S21). The estimates of ΔH, ΔS, and ΔG at pressures up to ∼200 MPa appear to be reliable within the experimental error, even if we consider ΔK and ΔE as p- and T-independent quantities.
In conclusion, the presented analysis shows that the compressibility ΔK and the expansibility ΔE are the key variables that determine the pressure dependence of the thermal stability of DNA. Using the thermodynamic equation of state and Maxwell’s relation, we predicted ΔK and ΔE as functions of temperature and pressure. These predictions suggest the existence of a convergence temperature, Tc, at which ΔV, dTm/dp, and ΔK simultaneously become equal to zero. While the observed volumetric and stability parameters differ between the duplexes and G-quadruplexes studied here, the slope (∂ΔK/∂T)p is very similar, likely due to a similar effect of temperature on nonspecific DNA hydration. We emphasized the importance of the hydration term as the dominant factor in defining the sign of ΔV and ΔK. At T < Tc the folded DNA structure is pressure unstable, since ΔV is negative (i.e., an increase in pressure leads to unfolding). At T > Tc ΔV becomes positive and the DNA structure is stabilized by an increase in pressure. Furthermore, our analysis suggests that Tc is pressure-independent. In this light, pressure induced destabilization and stabilization effects depend only on temperature and can be considered independent of the initial pressure at which the pressure perturbation is applied.
Overall, the thermodynamic analysis provides interesting new relationships between thermodynamic quantities that determine the pressure- and temperature-dependent changes in DNA stability.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Winter R. Interrogating the Structural Dynamics and Energetics of Biomolecular Systems with Pressure Modulation. Annu. Rev. Biophys. 2019, 48, 441–463. 10.1146/annurev-biophys-052118-115601.30943042 · doi ↗ · pubmed ↗
- 2Akasaka K.; Matsuki H.High Pressure Bioscience—Basic Concepts, Applications and Frontiers; Springer: 2015.
- 3Balny C.; Masson P.; Heremans K. High Pressure Effects on Biological Macromolecules: from Structural Changes to Alteration of Cellular Processes. Biochim. Biophys. Acta 2002, 1595, 3–10. 10.1016/S 0167-4838(01)00331-4.11983383 · doi ↗ · pubmed ↗
- 4Bartlett D. H. Pressure Effects on in vivo Microbial Processes. Biochim. Biophys. Acta 2002, 1595, 367–381. 10.1016/S 0167-4838(01)00357-0.11983409 · doi ↗ · pubmed ↗
- 5Daniel I.; Oger P.; Winter R. Origins of Life and Biochemistry under High-Pressure Conditions. Chem. Soc. Rev. 2006, 35, 858–875. 10.1039/b 517766 a.17003893 · doi ↗ · pubmed ↗
- 6Harrison J. P.; Gheeraert N.; Tsigelnitskiy D.; Cockell C. S. The Limits for Life under Multiple Extremes. Trends Microbiol 2013, 21, 204–212. 10.1016/j.tim.2013.01.006.23453124 · doi ↗ · pubmed ↗
- 7Meersman F.; Daniel I.; Bartlett D. H.; Winter R.; Hazael R.; Mc Millan P. F. High-Pressure Biochemistry and Biophysics. Rev. Mineral. Geochem. 2013, 75, 607–648. 10.2138/rmg.2013.75.19. · doi ↗
- 8Oger P. M.; Jebbar M. The Many Ways of Coping with Pressure. Res. Microbiol. 2010, 161, 799–809. 10.1016/j.resmic.2010.09.017.21035541 · doi ↗ · pubmed ↗