Potential roles of voltage-gated ion channel disruption in Tuberous Sclerosis Complex
Hailey X. Egido-Betancourt, Roy E. Strowd III, Kimberly F. Raab-Graham

TL;DR
This paper explores how disrupted voltage-gated ion channels may contribute to epilepsy in Tuberous Sclerosis Complex, a genetic disorder linked to mTOR signaling.
Contribution
The paper introduces a novel perspective on Tuberous Sclerosis Complex by linking mTOR signaling to voltage-gated ion channel dysfunction in epilepsy.
Findings
Tuberous Sclerosis Complex is associated with overactive mTOR signaling and a high incidence of epilepsy.
Voltage-gated ion channel mutations can alter neuronal excitability and cause seizures.
Emerging data suggests mTOR signaling may regulate voltage-gated ion channel expression in neurons.
Abstract
Tuberous Sclerosis Complex (TSC) is a lynchpin disorder, as it results in overactive mammalian target of rapamycin (mTOR) signaling, which has been implicated in a multitude of disease states. TSC is an autosomal dominant disease where 90% of affected individuals develop epilepsy. Epilepsy results from aberrant neuronal excitability that leads to recurring seizures. Under neurotypical conditions, the coordinated activity of voltage-gated ion channels keep neurons operating in an optimal range, thus providing network stability. Interestingly, loss or gain of function mutations in voltage-gated potassium, sodium, or calcium channels leads to altered excitability and seizures. To date, little is known about voltage-gated ion channel expression and function in TSC. However, data is beginning to emerge on how mTOR signaling regulates voltage-gated ion channel expression in neurons. Herein,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
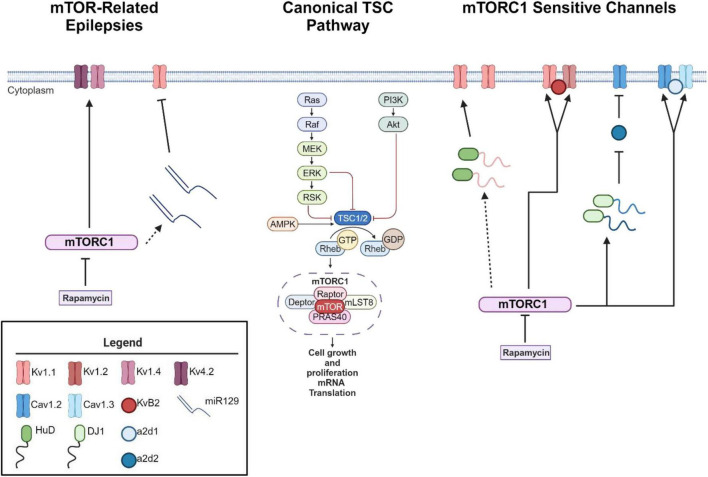 FIGURE 1
FIGURE 1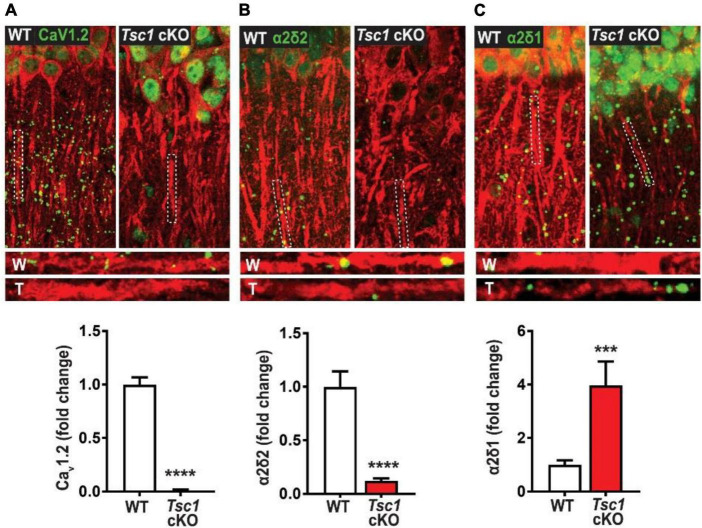 FIGURE 2
FIGURE 2| Gene name | Channel type | Localization | Seizure classification | Function | Direction noted | References |
|
| Kvβ1 | Brain | Early-onset epilepsy | Inactivation regulator of alpha potassium channels | Increase | |
|
| Cavβ2 | Cardiac, skeletal, smooth, and brain | Epilepsy | Modulation the gating of alpha calcium channels | Increase |
|
|
| Cavβ4 | Brain | Absence epilepsy; idiopathic generalized epilepsy; juvenile myoclonic epilepsy | Modulation the gating of alpha calcium channels | Increase |
| Gene name | Channel type | Localization | Seizure classification | Function | Observed expression | Effect on neuronal activity | References |
|
| Kv1.1 | Brain | Epilepsy; generalized or partial | Initiation and propagation, shaping, regulating action potential | Decrease | Increase | |
|
| Kv1.2 | Brain | Myoclonic epilepsy | Initiation and propagation, shaping, regulating action potential; Inactivation regulator of alpha potassium channels | Decrease | Increase | |
|
| Kv1.4 | Brain | Episodic Ataxia; Epilepsy | Regulates presynaptic neurotransmitter release; regulates intrinsic excitability | Decrease | Increase | |
|
| Kv4.2 | Brain | Infant-onset Epilepsy | Determine the extent of inactivation for the cell | Decrease | Increase | |
|
| Kvβ2 | Brain | Epilepsy | Inactivation regulator of alpha potassium channels | Decrease | Increase |
|
|
| Cav1.2 | Cardiac, smooth muscle, neuronal, adrenal, chromaffin cells | Febrile seizures | Regulates cardiac action potential; excitation-coupling | Increase (somatic); Decrease (dendritic) | Increase | |
|
| Cav1.3 | Endocrine, neuronal, adrenal, chromaffin cells | Epilepsy-Associated | Sinoatrial pacemaking | Increase | Increase | |
|
| α2δ1 | Skeletal muscle, brain | Epilepsy, cerebellar ataxia | Regulates VGCC current density, and activation/ | Increase | Increase | |
|
| α2δ2 | Lung, brain | Epileptic encephalopathy, ataxia | Regulates VGCC current density, and activation/ | Decrease | Increase |
| Gene Name | Channel type | Localization | Seizure classification | Function | Predicted channel function | References |
|
| Kv4.3 | Cardiac muscle, brain | Generalized epilepsy | Determine the extent of inactivation for the cell | GOF | |
|
| Cav2.1 | Neuronal | Absence seizures | Neurotransmitter release | LOF | |
|
| Cav2.3 | Neuronal | Absence epilepsy, human juvenile myoclonic epilepsy | Neurotransmitter release | GOF |
|
|
| Cav3.1 | Neuronal, cardiac | Absence seizures | Sleep regulation; pacemaking | GOF | |
|
| Cav3.2 | Neuronal, cardiac | Absence seizures | Regulation of neuronal firing; pacemaking activity | GOF | |
|
| Nav1.1 | Brain | Myoclonic epilepsy (Dravet syndrome); generalized epilepsy with generalized clonic seizures | Generation and propagation of action potentials | LOF |
|
|
| Nav1.2 | Brain | Atypical generalized epilepsy; febrile seizures | Generation and propagation of action potentials | GOF | |
|
| Nav1.3 | Brain | Cryptogenic partial epilepsy-associated; | Generation and propagation of action potentials | GOF and LOF | |
|
| Nav1.6 | Brain | Infantile epilepsy | Generation and propagation of action potentials | GOF |
|
|
| Nav1.7 | Brain | Febrile seizures; afebrile seizures, generalized tonic-clonic seizures, myoclonic or tonic seizures, focal clonic seizures | Generation and propagation of action potentials | GOF and LOF |
- —National Institute of Neurological Disorders and Stroke10.13039/100000065
- —National Institute on Alcohol Abuse and Alcoholism10.13039/100000027
- —Medical Research and Materiel Command10.13039/100000182
- —National Institute on Drug Abuse10.13039/100000026
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTuberous Sclerosis Complex Research · Ion channel regulation and function · Polyomavirus and related diseases
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant disease affecting roughly 1 in 6000 live births, with an estimated prevalence of 1 in 14,000 to 1 in 25,000 (Curatolo and Moavero, 2012; Kothare Sanjeev et al., 2014). Disease causing mutations in either the TSC1 or TSC2 gene lead to loss of protein function (O’Callaghan et al., 2004; Curatolo et al., 2008). TSC1 and TSC2 form dimers to inhibit the activity of mammalian target of rapamycin (mTOR), composed of two complexes mTORC1 and mTORC2. Loss of either TSC1 or TSC2 leads to hyperactive mTOR signaling and tuberous malformations. More than 90% of affected individuals experience seizures over the course of their lifetime (O’Callaghan et al., 2004; Kelleher and Bear, 2008; Stafstrom et al., 2017). As the etiology as TSC is well established, mechanism based treatments such as the mTORC1 inhibitor, rapamycin and other “rapalogues”, has been the focus of several clinical trials to treat TSC-related seizures (Franz et al., 2018).
TSC patients suffer from both focal and generalized epilepsy syndromes including febrile seizures, infantile spasms, focal seizures, and absence seizures (Kothare Sanjeev et al., 2014). Seizures may arise in TSC in two possible ways. Some studies speculate that seizure activity is generated by brain malformations that result from cortical tubers, which are composed of dysmorphic neurons and gliotic cells and are commonly seen in TSC patients (Stafstrom et al., 2017; Zou et al., 2017). The tubers may be surgically removed to provide temporary relief from the seizures (Bollo et al., 2008). Second, some studies suggest that hyperactive mTOR signaling itself can disrupt the excitatory/inhibitory (E/I) balance among neuronal networks (Bateup et al., 2013). Thus, in the absence of tubers, TSC patients may be susceptible to seizure-like activity and downstream neuronal damage due to hyperactive mTORC1 signaling, further disrupting mRNA translation and protein expression (Bateup et al., 2013). Cortical tuber development has been widely studied in TSC [previously reviewed in Wong (2008) and Lu et al. (2018)]; however, little is known regarding the molecular underpinnings of hyperexcitable networks downstream of mTOR signaling, that underlie epilepsy in TSC, in the absence of cortical tubers.
For decades, dysregulation of ion channels, such as voltage-gated potassium, calcium, and sodium channels, has been suggested to be the leading cause of shifts in neuronal excitability, that underlie epilepsy (Poolos and Johnston, 2012). Recently, evidence linking mTOR signaling to ion channel expression in neurons has emerged [reviewed further in Raab-Graham and Niere (2017)]. Together, these findings have led us to ask the question of whether voltage-gated ion channels are contributing to TSC-related seizures. Herein, we discuss known voltage-gated ion channels currently associated with acquired epilepsies, but not yet understood in the context of TSC. The goal of this review will be to extrapolate and expand on the current findings of voltage-gated channels implicated in other epilepsies, where aberrant mTOR signaling occurs, while surmising their role in TSC-related seizures.
mTOR as a putative voltage sensor
As mentioned above, loss of function mutations in the TSC1 or TSC2 genes results in hyperactive mTOR signaling (Curatolo et al., 2008; Cho, 2011; Wong, 2013; Stafstrom et al., 2017). mTOR consist of two protein complexes, mTORC1 and mTORC2. Herein, we will focus on mTORC1 signaling as it is a serine/threonine kinase that regulates mRNA translation (Curatolo et al., 2008; Curatolo and Moavero, 2012; Curatolo et al., 2015; Roach, 2016; Raab-Graham and Niere, 2017), which may alter the expression of epilepsy associated ion channels in neurons (Figure 1). mTORC1’s downstream signaling is required for many forms of synaptic plasticity, synapse formation, and recently ? Site specific expression of ion channels in neuronal dendrites (Raab-Graham et al., 2006; Cho, 2011; Brewster et al., 2013; Meng et al., 2013; Wong, 2013). Thus, an emerging theory is that mTORC1 activity may serve as a “voltage-sensor” turning on and off to maintain the membrane potential in an optimal range, through protein synthesis and repression of ion channel mRNAs (Figure 1; Niere and Raab-Graham, 2017). Thus, if mTOR activity is constitutive, as in the case of TSC, ion channel expression/repression that promotes neuronal excitability will go unchecked (Lasarge and Danzer, 2014). Epilepsy has been classically considered a disorder of ion channel dysfunction (Poolos and Johnston, 2012). Together, these data may explain why independent studies suggest that overactive mTOR signaling itself can lead to epilepsy (Zeng et al., 2008; Curatolo et al., 2015; Sosanya et al., 2015; Niere and Raab-Graham, 2017). To date, the literature is sparse in its consolidation of excessive mTOR signaling and ion channel dysfunction in TSC-related epilepsies.
mTORC1 signaling leads to changes in ion channel expression. Schematic of canonical mTOR signaling pathway (middle). mTORC1 signaling pathway represses the mRNA translation of potassium channels such that mTORC1 inhibition with the drug rapamycin increases the expression of Kv1.1, Kv1.2, and Kvβ2 (right) (Sosanya et al., 2013, 2015; Niere and Raab-Graham, 2017). Interestingly, mTOR hyperactivity differentially alters calcium influx via Cav1.2, α2δ2, and Cav1.3 channel expression (Hisatsune et al., 2021; Niere et al., 2023). mTOR hyperactivity leads to augmented calcium influx in the soma via an increase in Cav1.3 gene and protein expression and slight increase of Cav1.2 (Hisatsune et al., 2021) (right). On the other hand, RNA binding protein DJ1 binds the mRNA coding for Cav1.2 and α2δ2 and represses translation. This repression causes deficits in L-VGCC dendritic signaling (Niere et al., 2023). Notably, with seizure induction, blocking early-stage epileptogenesis in a model of temporal lobe epilepsy with rapamycin increases the expression of Kv1.1 (left). A secondary mechanism of repression kicks in to repress Kv1.1 expression with extended use of rapamycin (Sosanya et al., 2015) while continued use of rapamycin restores Kv1.4 and Kv4.2 (Brewster et al., 2013) during late stage epileptogenesis. Dynamic expression of RNA-binding proteins and microRNAs regulate the expression of Kv1.1. HuD increases the translation of Kv1.1 when mTORC1 is turned off and miR-129 represses the translation of Kv1.1 when mTORC1 is turned on. For further mechanistic detail, please see the following articles: (Sosanya et al., 2013, 2015). Interestingly, miR-129 expression increases with extended use of rapamycin, likely drives the second wave of Kv1.1 repression and an increases in seizure frequency (Sosanya et al., 2015). Dotted lines reflect multiple molecular steps between proteins. Created with BioRender.com. Agreement number: VB26LQZB9H.
Types of TSC-associated seizures
The most common type of seizure in children with TSC are infantile spasms, occurring between 3 and 9 months after birth (Pellock et al., 2010; Appleton, 2011; Randle, 2017). Many different semiologies can be observed such as eye deviation as well as sudden bilateral and symmetrical tonic contractions, which last a few seconds (Curatolo et al., 2001). It has long been hypothesized that if these seizures are left untreated, children suffering from infantile spasms will experience impairment in developmental progress and more severe neurologic problems, such as autism spectrum disorder (Pellock et al., 2010, Alliance, 2020). Interestingly, several clinical trials have since discovered contrary findings with respect to targeting early life infantile spasms in TSC patients. One such trial found that preventative treatment with vigabatrin, the first line of treatment for infantile spasms which targets gamma-amino butyric acid (GABA)-transaminase, ultimately increases the concentration of GABA present in the brain (Yum et al., 2013). However, treatment did not delay or lower the incidence of other seizure types, such as focal and drug resistant epilepsy nor improve neurocognitive outcome at 24 months of age in TSC children (Bebin et al., 2024). On the other hand, TSC patients who showed signs of epileptiform activity before seizure onset, and were treated with vigabatrin, took longer to display clinical seizure and the preventative treatment reduced the risk of other clinical seizures (Kotulska et al., 2021); however, neurocognition was not determined. Further research targeting the underlying mechanisms of infantile spasms.
The second most common seizure type is focal onset seizures, previously called focal or partial seizures, as they originate at some specific point in the brain. These seizures differ from infantile spasms in that they can be either awareness or impaired awareness with non-motor onset or motor onset (Almobarak et al., 2018). These seizures can precede or coexist with infantile spasms, or even evolve from infantile spasms (Yum et al., 2013). While the cause of focal seizures is not fully understood, some suggest that focal insults are caused by brain malformations resulting from structural tuber alterations (Stafstrom and Carmant, 2015; Curatolo et al., 2018). To mimic focal seizures in a mouse model of TSC, pups in utero underwent electroporation to focally express constitutively active Rheb (Rheb*^CA^), to augment mTOR activity only in the selected area. Indeed, this model is similar to a model of cortical dysplasia that experiences focal seizures as a result of expressing Rheb^CA^* (Hsieh et al., 2016). The authors found that varying the concentration of Rheb led to high levels of mTOR activity, which increased seizure frequency and correlated with the degree of disease severity (Nguyen et al., 2019). These findings further the notion that focal seizures, in the absence of tuber abnormalities, is thought to be caused by select mTOR-afflicted neurons, and results in altered network excitability and seizures.
TSC patients may also suffer from generalized onset, formerly known to encompass tonic seizures, myoclonic seizures, and absence seizures (Appleton, 2011; Kiriakopoulos and Osborne, 2017). These seizures can begin focally and bilaterally expand to larger aspects of the cortex, although not necessarily the entire cortex. Patients with generalized onset seizures present with stiffened muscles, rhythmical jerking, and impaired awareness (Stafstrom and Carmant, 2015, Almobarak et al., 2018).
There is a substantial portion of TSC patients that continue to have seizures despite maximum aggressive anti-seizure treatment. For these patients, new approaches to management are needed. For example, if focal seizures coexist or precede infantile spasms, vigabatrin treatment can be less effective or have no effect. This is thought to be due to the medication only targeting one seizure type (Yum et al., 2013). Thus, it is imperative to determine the “molecular origin” of seizure onset, in order to better determine the course of treatment for a TSC affected individual.
Clues from transcriptome studies of TSC, mTOR-mediated ion channel Expression, and speculated epilepsy
A few studies have examined the transcriptome of human cortical tubers removed from patients with TSC (Boer et al., 2010; Mills et al., 2017). Table 1 lists transcripts associated with voltage-gated channel expression (Heinemann et al., 1996; Escayg et al., 1998; Ebert et al., 2008; Zhang et al., 2015). Interestingly, all the genes listed code for auxiliary subunits that serve to increase the surface expression of the pore-forming subunit or change the ion channel kinetics. These data further convey the need to investigate the expression and function of the pore forming subunits in neuronal models of TSC. While mRNA does not necessarily mean changes in protein expression, others have demonstrated that mTOR is overactive in other models of epilepsy, similar to TSC, and these genes that code for these mTOR-dependent ion channels are summarized in Table 2 (Wang et al., 1993; McCormack et al., 1995; Burkhalter et al., 2006; Imbrici et al., 2007; Christel et al., 2012; Lee et al., 2014; Xie et al., 2014; Villa and Combi, 2016; Punetha et al., 2019; Dahimene et al., 2022). Finally, in Table 3, we propose a list of putative voltage-gated ion channels that may be dysregulated in TSC (Serôdio and Rudy, 1998; Wappl et al., 2002; Jarnot and Corbett, 2006; Ogiwara et al., 2007, 2018; Estacion et al., 2010; Smets et al., 2015; Wang et al., 2016; Wormuth et al., 2016; Fan et al., 2017; Zhang et al., 2020). Although, currently, there is no direct evidence of the potential role of dysfunction in the following voltage-gated ion channels leading to the hyperexcitable pathology in TSC, we speculate that these channels may play a role in the different seizure types present in TSC patients throughout their lives.
Voltage-gated potassium channels
Voltage-gated potassium (K_v) channels represent the largest family of genes in the Kv_ channel family that set the resting membrane potential and repolarize action potentials (Cooper, 2012; Robbins and Tempel, 2012). In general, K_v_ channels dampen neuronal activity, so loss of function (LOF) mutations lead to hyperexcitabile circuits and seizures. Potassium channels have different family subtypes that have distinct but similar function. The potassium channel consists of four α subunits and can include four cytoplasmic auxiliary β subunits (Robbins and Tempel, 2012:1). The different configurations of α and β subunits create different properties that dictate their biophysical properties including voltage-sensing and gating properties, described below.
Kv1
KCNA1 (K_v1.1) and KCNA2 (Kv1.2) have been associated with epilepsy (Cooper, 2012; Robbins and Tempel, 2012; Boutry-Kryza et al., 2015). The Kcna family codes for the pore-forming “Kv1” subunits, which may compose either A-type or delayed rectifier channels (Raab-graham and Niere, 2017). A-type currents are rapidly activating and fast inactivating, while delayed rectifiers open slowly and remain open (Raab-Graham and Niere, 2017). Depending on the brain region and cellular composition, the most abundantly expressed α subunits of the Kv1 subfamily are Kv1.1, Kv1.2, and Kv1.4 (Robbins and Tempel, 2012:1). Interestingly, Kv1.1 codes for a delayed rectifier, while Kv1.4 codes for the A type family. However, Kv1.1 in conjunction with a Kvβ1 or Kv1.4 subunit, can have properties of the A type family (D’Adamo et al., 2020). Kv1 channels are responsible for resetting the resting membrane potential and titrating synaptic release in neurons (Foust et al., 2011; Robbins and Tempel, 2012). Interestingly, reduced expression of either Kv1.1 and Kv1.2 channels have been associated with epilepsy, as each knockout mouse presents with seizures that resemble the development of human epilepsy (Robbins and Tempel, 2012). Additionally, mutations in Kv_ channels that disrupt coassembly with other α or β subunits reduces channel functional and/or expression (D’Adamo et al., 2020). Together, Kv1 channels prevent “runaway” depolarization, increased firing rates, and excessive neurotransmitter release, all of which can lead to seizures.
It should be noted, that cross referencing the transcriptome of human TSC cortical tubers and those genes with associated epilepsies, transcripts coding for K_v_ auxiliary subunits K_vβ1 and Kvβ2 were detected (Tables 1, 2; Boer et al., 2010). Interestingly, other Kv_ subunits such as K_v1.1, Kv1.2, Kv1.4, and Kv4.2 in have been implicated in mTOR-related epilepsy models (Table 2; Brewster et al., 2013; Niere and Raab-Graham, 2017). Together, these data suggest that the Kv_1 class should be further investigated in TSC.
Kv4
Among the K_v4 subunits, KCND2 (Kv4.2) and KCND3 (Kv4.3) also belong to A-type voltage-gated potassium channel class. These channels are abundantly found in the nervous system within somatodendritic compartment of neurons (Zemel et al., 2018). Interestingly, several studies have shown that down regulation of the A-current leads to increased excitability (Bernard et al., 2004; Liu et al., 2014). For example, a mutation in Kv4.2 (V404M) leads to impairments in inactivation after channel opening. This mutation has been associated with infant-onset epilepsy and autism (Lin et al., 2018). Thus, examining the Kv_4 subunit class maybe be a potential interest to TSC and associated seizure types. Altogether, we predict that these channels to be dysfunctional in TSC, specifically the potassium genes listed in Tables 1–3.
The role of mTOR in regulating Kv channel expression
Since the discovery of on demand local protein synthesis occurring at the synapse, dysregulation of protein synthesis can lead to misexpression of ion channel subunits and alter the membrane potential and consequently lead to seizure-like conditions (Switon et al., 2017). Pathways involving mTOR, are known to regulate local synthesis at the synapse. Under conditions where mTOR is active, local synthesis of K_v1.1 is repressed on dendrites without altering axonal expression (Raab-Graham et al., 2006; Niere and Raab-Graham, 2017). Additionally, others have shown that both Kv1.2, and Kv_β2 at the synapse are reduced when mTOR is active (Niere and Raab-Graham, 2017). Together, these findings suggest that mTOR activity toggles expression of potassium channels as a local feedback mechanism that ensures optimized synaptic function (Raab-Graham and Niere, 2017).
If mTOR activity is left unregulated, as seen in TSC, repression of K_v_ channel expression may lead to an increase in neuronal excitability and to eventual epileptogenesis (Cho, 2011; Meng et al., 2013; Jeong and Wong, 2016). This is suggested by Brewster and colleagues who utilized a model of pilocarpine-induced status epilepticus (SE) model and examined ion channel expression in presence and absence of rapamycin, an mTOR inhibitor. With the development of epileptogenesis, reduced expression of K_v1.4 and Kv4.2 in the hippocampus was observed. With the addition of rapamycin, which has been shown to reduce seizure frequency (Zeng et al., 2009) in SE rodents, protein levels of Kv1.4 and Kv4.2 increases to similar levels as seen in the vehicle treated rodents (Brewster et al., 2013). Altogether, these independent studies indicate that alterations in Kv_ expression could result in altered neuronal excitability and further studies are needed to implicate K_v_ channels to seizures such as those experienced in TSC.
Voltage gated sodium channels
Voltage-gated sodium channels are responsible for the generation and the propagation of action potentials along nerve cells (Catterall, 2000; Catterall et al., 2005). Mutations in sodium channel genes most commonly augment neuronal excitability leading to epilepsy (Menezes et al., 2020). The sodium channel is a transmembrane channel consisting of an α subunit and an auxiliary β subunit (Yu and Catterall, 2003; Mantegazza and Catterall, 2012). The α subunit contains four homologous domains composed of a voltage-sensing component and a pore-forming component which undergoes modifications by the auxiliary β subunit (Catterall, 2000; Catterall et al., 2005). There are nine sodium channel isoforms; however, only the sodium channels directly implicated in excitability will be mentioned here (Table 3), and described below.
Nav1.1 and Nav1.2
Of interest are Na_v1.1 and Nav1.2, channels expressed in neurons, but more specifically the gene mutations affecting the α subunits of these channels. These mutations lead to inherited forms of epilepsy that differ based on type of α subunit defect (Mantegazza and Catterall, 2012). The SCN1A gene, which encodes the Nav1.1 channel, has been associated with Dravet syndrome, which displays afebrile intractable seizures (Craig et al., 2012; Schmunk and Gargus, 2013). Missense mutations in the SCN1A gene (D322N), commonly display gain of function (GOF), that lead to enhanced sodium currents as a result of lack of inhibition on excitatory neurons (Menezes et al., 2020). Likewise, mutations in the SCN2A gene (A467T), that encodes the voltage-gated sodium channel Nav_1.2, have been shown to elicit seizure behavior such as in generalized epilepsy with febrile seizure plus (GEFS+) syndrome by also enhancing sodium currents (Schmunk and Gargus, 2013; Wolff et al., 2017). Additionally, LOF mutations in SCN2A have been linked to ASD and intellectual disability, all of which are commonly seen in patients with TSC. Thus, further research is needed to understand the mechanisms by which mutations in these genes leads to TSC.
Nav1.3, Nav1.6, and Nav1.7
Other voltage-gated sodium channels, such as Na_v1.6 and Nav1.7 are associated with infantile spasms and febrile seizures, respectively, while Nav1.3 has been associated with patients with epilepsy (Menezes et al., 2020). Notably, mutations in SCN8A gene, coding for Nav1.6, affects the action potential threshold which increases spontaneous and repetitive firing leading to an increase in excitability (Menezes et al., 2020). Additionally, GOF and LOF mutations in Nav1.3 and Nav1.7, have been reported to alter the biophysical properties of neurons as these genes modify other sodium channels such as Nav_1.1, which can contribute to pathogenesis of epilepsy, whoever, more studies are needed to ascertain their direct involvement. Altogether, these channels are associated with types of seizures experienced within TSC, however, these channels remain uninvestigated, making these channels possible candidates to examine in TSC.
Voltage-gated calcium ion channels
Speculated voltage-gated ion channels in TSC associated epilepsy
Voltage-gated calcium channels are required for different functions in the neuron, such as controlling neuronal excitability and regulating calcium-sensitive intracellular pathways (Cain and Snutch, 2012; Zamponi et al., 2015). There are three classes of voltage-gated calcium channels, Ca_v1, Cav2, and Cav_3 (Table 3). Each class has subclasses of ion channel expression that vary based function, and kinetics. The channels are either high voltage (HVA) or low voltage activated (LVA), meaning the channel opens or activates at −40 and −60 mV, respectively (Cain and Snutch, 2012). The calcium channel, like the sodium and potassium channel, contain an α subunit that stands as the pore-forming unit that is selective for calcium (Cain and Snutch, 2012; Zamponi et al., 2015). They also have auxiliary subunits β, γ and α2δ that regulate the properties of the channel (Campiglio and Flucher, 2015). Because of the genetic diversity among the calcium channels, only a select few of the channels expressed in the brain will be discussed, specifically the HVA and LVA channel subunits listed in Tables 1–3.
The β auxiliary subunits play an important role in enhancing the biophysical properties of the α subunit, such as channel folding, channel trafficking, and alters gating kinetics and voltage-dependence (Dolphin, 2003, 2016). Interestingly, our comparison of the human TSC cortical tuber transcriptome cross referenced with genes associated with epilepsies, yielded elevated mRNA coding for Ca_vβ2 and Cavβ4 (Table 1; Boer et al., 2010). Interestingly, one study demonstrated that ablation of Cavβ1, Cavβ2, and Cavβ3 have no major impact on neuronal function (Ball et al., 2002; Vergnol et al., 2022). On the other hand, one study demonstrated Cavβ4 is associated with the lethargic mouse model of epilepsy (Burgess et al., 1997; Vergnol et al., 2022), while another study showed that disruption in the Cav_β2 gene leads to diminished L-type channel currents (Weissgerber et al., 2006). Although the biophysical properties of these two subunits have yet to be determined in TSC, this finding shows possible insights into voltage-gated calcium channels and whether they are disrupted.
Cav2
The Cav2 family encompasses Ca_v2.1, Cav2.2, and Cav2.3 isoforms. these channels are comprised of a pore-forming α subunit and auxiliary β subunits. Together, they are responsible for regulating Ca2+ entry in response to depolarization and release of neurotransmitters (Mochida, 2019). These channels can undergo alternative splicing, and thus, have a wide spectrum of biophysical properties. Of particular interest are Cav2.1 and Cav2.3, as shown in Table 3, will be discussed further as these channels have more direct implications to seizure. There are several Cav2.1 channel mutations that generate epileptic phenotypes commonly seen within TSC. For example, TSC patients can suffer from absence epilepsy, whose mouse models, “leaner”, “tottering”, and “rocker,” display epileptic phenotypes as a result of different Cav2.1 channel mutations (Imbrici et al., 2004; Mochida, 2019). These mutations affect Cav2.1 current density by slowing channel inactivation as well as imbalances on inhibitory to excitatory neurotransmission leading to increased firing (Rajakulendran and Hanna, 2016). Similarly, Cav2.3 has also been demonstrated to play a role in absence epilepsy role (Zaman et al., 2011). Nevertheless, the contribution of Cav2.1 or Cav_2.3 to TSC absence epilepsy remains to be determined and, thus, this remains a possible avenue of exploration.
Cav3
T-type calcium channels, do not require auxiliary subunits (Simms and Zamponi, 2014). Because the Ca_v3 subunits have been shown to undergo alternative splicing, resulting in channel function diversity, the T-type channels that will be discussed in the context of TSC will be Cav3.1 (CACNA1G) and Cav3.2 (CACNA1H). CACNA1G channels are highly expressed in thalamocortical (TC) neurons (Chen et al., 2014). CACNA1H has been shown to be primarily expressed in the dorsal root ganglion, dentate of the hippocampus, and thalamus (Graef et al., 2011; Simms and Zamponi, 2014; Bernal Sierra et al., 2017). Cav_3.2 mutations have been shown to lead to seizures in murine models, specifically absence epilepsy. Because of this implication in seizure commonality, and because this limbic seizure can precede from subcortical structures such as the thalamus, it is possible that T-channels play a role in initiating the spread to higher structures in the TSC brain.
CaV channel expression in TSC
As previously mentioned above, the HVA class also encompasses L-type calcium channels as they are integral to cell’s membrane complex that mediate influx of Ca2+ after a depolarization response (Hofmann et al., 2014). The “L” in L-type represents the long-lasting inward currents during depolarization which have distinguished them from their “transient current” T-type cousins (Zamponi et al., 2015). One L-type channel, Ca_v1.2, is composed of three subunits α1, α2δ, and β axillary subunits (Hofmann et al., 2014; Zamponi et al., 2015), and may provide more insight into TSC epileptic phenotypes. There have been case studies indicating the presence of febrile seizures among TSC patients (Kubo et al., 2011; Siddaraju et al., 2016). However, these case studies primarily served as documentation for the patients’ condition, and no other studies have followed up on the possibilities of febrile seizures in TSC models. Interestingly, one independent study demonstrated that febrile seizures in rat pups may be prevented with the use of nimodipine (Radzicki et al., 2013). Additionally, mTOR hyperactivation has been shown to differentially regulate L channel expression in TSC. An independent study has demonstrated that somatic Cav1.2 and Cav1.3 gene and protein expression are augmented in TSC2-null neurons. Hisatsune et al., 2021 also demonstrates that Cav1.3 triggers enhanced neuronal activity of TSC2^–/–^ neurons and could be a potential novel target for epilepsy in TSC (Hisatsune et al., 2021). On the other hand, Cav1.2 de novo protein synthesis was found to be reduced in the dendrites of hippocampal CA-1 neurons in a mouse model of TSC1 (Figure 2). Furthermore, Niere et al. (2023) found that an RNA binding protein DJ-1 coordinates the expression of Cav_ channel complex, including Cav1.2 and α2δ2, resulting in attenuated calcium signaling in the dendrites (Figure 2 and Table 2). Like the β subunits mentioned above, the α2δ auxiliary subunits play an important role in trafficking and gating of the α subunits (Dolphin, 2003, 2016). Additionally, α2δ1 was found to be overexpressed in conditionally knockout TSC hippocampal dendrites (Niere et al., 2023); however its role in TSC has not been established. Together, these two studies on L-type calcium channels, suggest that subcellular localization of these channels differentially affects calcium influx across the cell. Considering the importance of calcium channel to seizures, these findings give credence to further investigation of calcium channel dysfunction in TSC.
*Tsc1 cKO mouse model exhibits decreased de novo protein synthesis of CaV1.2 α2δ2, but not α2δ1 (40). De novo protein synthesis, visualized by Surface Sensing of Translation-Proximity Ligation Assay (SUnSET-PLA) (green) in hippocampal dendrites (MAP2, red). The SUnSET-PLA combinatory assay labels newly synthesized CaV1.2, α2δ2, and α2δ1 proteins in the hippocampus by detecting puromycin (which binds and halts translation) on a translating ribosome and the translating protein with a specific antibody. This assay allows one to separate new protein from already synthesize protein. WT (W) and TSC (T) dendrites are outlined by broken lines. (A) Basal CaV1.2 protein synthesis is detected in dendrites of WT is markedly reduced in TSC. (B) α2δ2 basal new protein synthesis is detected in dendrites of WT but is attenuated in TSC. (C) Basal α2δ1 protein synthesis in dendrites of WT is lower than TSC. For representative images in panel (A) through panel (C), CaV1.2, α2δ2, and α2δ1 puncta were dilated once using ImageJ. Adapted from Niere et al. (2023). Bar values represent mean ± SEM. ***P < 0.001, ***P < 0.0001.
Conclusion
In conclusion, the disruption of voltage-gated ion channels leads to different types of seizures. mTOR, downstream of the TSC, has been shown to be involved in regulating ion channel expression, and may contribute to epileptogenesis. Because of the complexity each voltage-gated ion channel, there are many unanswered questions of their role in TSC. Yet, understanding the contribution from each voltage-gated ion channel may provide insight into the heterogeneity of seizures in TSC and possibly determine new therapeutic targets of interest.
Scope statement
Tuberous sclerosis complex (TSC) is a neurodevelopmental disorder that results in hyperactive mammalian/mechanistic target of rapamycin (mTOR) signaling leading to altered neuronal excitability and seizures; however, the underlying mechanisms remain a mystery. Several potassium, sodium, or calcium voltage-gated channels have been found to be causative in disorders that result in aberrant neuronal excitability and seizures. Surprisingly, these channels remain understudied in TSC-associated neuronal dysfunction. The coordination of these ionic conductances, dictated by the channel’s expression, subcellular localization, and biophysical properties, keep neurons operating in an optimal range, providing network stability. Our review examines the current TSC literature describing common seizure types, clinical trials, and genomic studies that potentially implicate potassium, sodium, and calcium voltage-gated channel dysfunction in TSC. Notably, the expression of several voltage-gated ion channels and auxiliary subunits have been shown to be regulated by mTOR signaling, arguing for further studies of ion channel dysfunction in TSC. Frontiers in Molecular Neuroscience-Molecular Signalling and Pathways is particularly interested in topics that pertain to brain disease mechanisms such as TSC, molecular signaling pathways such as mTOR, and synaptic and cellular proteins such as voltage-gated ion channels.
Data availability statement
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
HE-B: Conceptualization, Data curation, Writing – original draft, Writing – review and editing. RS: Writing – review and editing. KR-G: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review and editing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alliance T. S. (2020). Infantile spasms [Online]. Available online at: https://www.tsalliance.org/about-tsc/signs-and-symptoms-of-tsc/brain-and-neurological-function/infantile-spasms/ (accessed October 06, 2020).
- 2Almobarak S.Almuhaizea M.Abukhaled M.Alyamani S.Dabbagh O.Chedrawi A. (2018). Tuberous sclerosis complex: Clinical spectrum and epilepsy: A retrospective chart review study. Transl. Neurosci. 9 154–160.30479846 10.1515/tnsci-2018-0023 PMC 6234476 · doi ↗ · pubmed ↗
- 3Appleton R. E. (2011). Tsc and epilepsy [Online]. Available online at: https://tuberous-sclerosis.org/wp-content/uploads/2019/10/TSA-TSC-and-epilepsy.pdf (accessed October 6, 2020).
- 4Ball S. L.Powers P. A.Shin H. S.Morgans C. W.Peachey N. S.Gregg R. G. (2002). Role of the beta(2) subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest. Ophthalmol. Vis. Sci. 43 1595–1603.11980879 · pubmed ↗
- 5Bateup H. S.Johnson C. A.Denefrio C. L.Saulnier J. L.Kornacker K.Sabatini B. L. (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78 510–522. 10.1016/j.neuron.2013.03.017 23664616 PMC 3690324 · doi ↗ · pubmed ↗
- 6Bebin E. M.Peters J. M.Porter B. E.Mcpherson T. O.O’kelley S.Sahin M. (2024). Early treatment with vigabatrin does not decrease focal seizures or improve cognition in tuberous sclerosis complex: The prevent trial. Ann. Neurol. 95, 15–26.10.1002/ana.26778 PMC 1089952537638552 · doi ↗ · pubmed ↗
- 7Bernal Sierra Y. A.Haseleu J.Kozlenkov A.Bégay V.Lewin G. R. (2017). Genetic Tracing of Ca(v)3.2 T-type calcium channel expression in the peripheral nervous system. Front. Mol. Neurosci. 10:70. 10.3389/fnmol.2017.00070 28360836 PMC 5350092 · doi ↗ · pubmed ↗
- 8Bernard C.Anderson A.Becker A.Poolos N. P.Beck H.Johnston D. (2004). Acquired dendritic channelopathy in temporal lobe epilepsy. Science 305 532–535.15273397 10.1126/science.1097065 · doi ↗ · pubmed ↗