Erdheim-Chester Disease Presenting as Bilateral Facial Masses: A case report and review of literature
Asim Qureshi, Abdulaziz Bakathir, Fizza Qureshi, Amanullah Beg, Asem Shalaby

TL;DR
A rare case of Erdheim-Chester disease in Oman is reported, marked by recurring facial masses and multiple health complications.
Contribution
This is the first documented case of Erdheim-Chester disease in Oman with a focus on recurrent bilateral facial masses.
Findings
The patient exhibited bilateral facial masses that recurred over 10 years.
Biopsy confirmed classic features of ECD, including foamy histiocytes and Touton giant cells.
The patient also developed xanthelasmas, bone lesions, and endocrine complications.
Abstract
Erdheim-Chester disease (ECD) is a rare form of non-Langerhans cell histiocytosis with unknown aetiology. It is recently recognised to be neoplastic with genetic mutations affecting the mitogen-activating protein kinase pathway. We report a 49-year-old female patient who initially presented in 2012 to a tertiary care centre in Muscat, Oman, with bilateral facial masses. These were removed but later recurred over a period of 10 years. She then presented with xanthelasmas, bone lesions, secondary infertility due to hypothalamic hypogonadism, diabetes insipidus and Hashimoto’s hypothyroidism. The facial masses were biopsied and they showed classic morphological features in the form of diffuse infiltration by foamy histiocytes with scattered Touton type of giant cells, patchy lymphocytic infiltrates and dense fibrosis. The patient is stable and is being followed-up. The presented ECD case…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
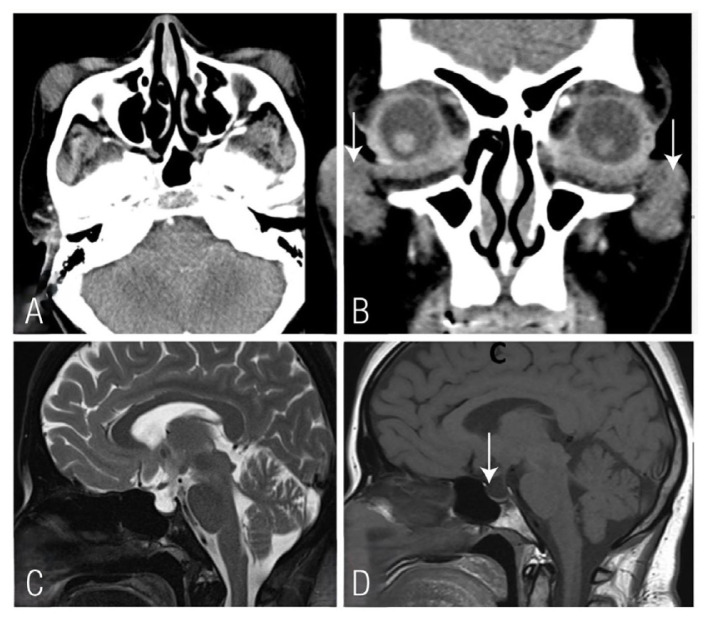 Figure 1
Figure 1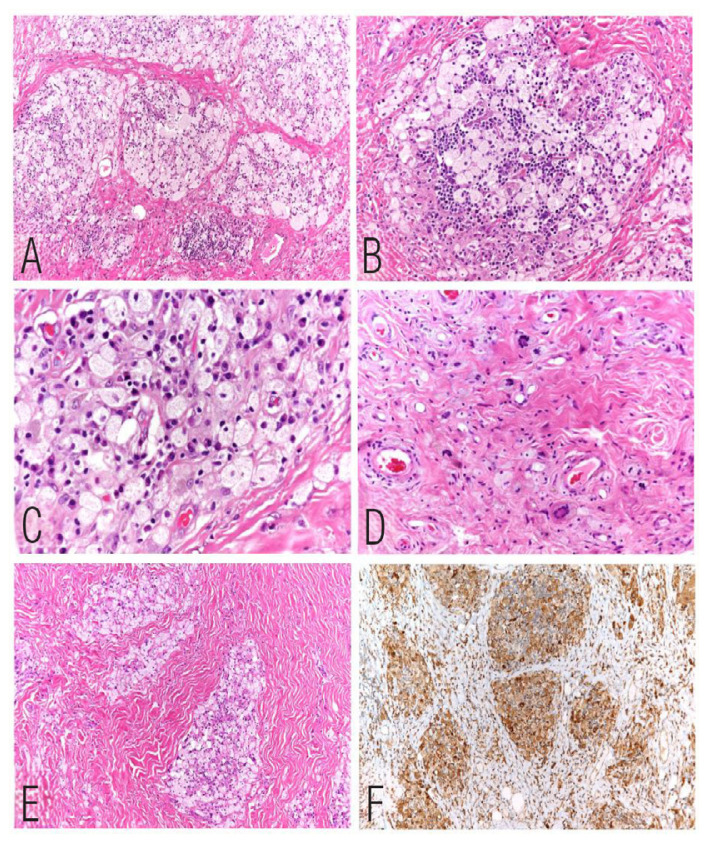 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpilepsy research and treatment · Pharmacological Effects and Toxicity Studies · Neonatal and fetal brain pathology
Erdheim-Chester disease (ECD) is a rare non-Langerhans cell histiocytic disease. It was first described in 1932 by William Chester while he was visiting Jakob Erdheim in Vienna, Austria. They reported the disease as “lipoid granulomatosis”.1 In 1972 Dr. Ronald Jaffe reported a third similar case and coined the name ECD.2 It is a rare disease that is being increasingly recognised; more than 1,000 cases have been reported in the literature over the last decade.3
This rare, potentially fatal multi-organ myeloid neoplasm occurs primarily in adults with a slight male predominance. It is believed to be under-diagnosed due to the wide variety of manifestations often mimicking other diseases and simultaneous involvement of multiple organ systems.4 The mean age of onset is 55–60 years. Clinical presentation varies depending on the extent and distribution of the disease and may range from asymptomatic incidentally diagnosed bone lesions to multisystemic, life-threatening forms with poor prognosis.
The diagnosis of ECD is based on a combination of histopathological, clinical and radiological features. It is reported that more than 95% of ECD patients have skeletal involvement. The commonest of which is bilateral and symmetric cortical osteosclerotic lesions of the diaphyseal and metaphyseal regions of the long bones especially the distal femur, proximal tibia and fibula. These radiological characteristics are highly suggestive of the disease.5 The extraskeletal manifestations may include exophthalmos, diabetes insipidus, interstitial lung disease, cardiovascular involvement, adrenal enlargement, retroperitoneal fibrosis, renal impairment, testis infiltration, breast involvement and central nervous system manifestations.6 A tissue biopsy is mandatory for histological confirmation and molecular profiling is required for therapeutic purposes. It is a clonal disorder associated with mitogen-activating protein kinase (MAPK) and BRAF V600E mutations.7
Case Report
A 49-year-old female presented to a tertiary care centre in Muscat, Oman, in 2012 with bilateral facial swellings. The right-side lesion was removed and histopathology showed a fibrohistiocytic lesion with foam cells. This was diagnosed as a benign fibrohistiocytic lesion. Later, she presented to the outpatient department with multiple other problems including fertility issues. She is known to have hypothalamic hypogonadism, diabetes insipidus, Hashimoto’s hypothyroidism, xanthelasmas and was recently diagnosed to have systemic lupus erythematosus (SLE). The facial lesion on the right side began to reappear and was again the size that was previously removed. A computed tomography scan of the head and neck showed bilateral soft tissue density masses in pre-zygomatic areas with no calcification or underlying bony erosions [Figure 1]. The plain radiography of the left lower limb showed sharply circumscribed sclerotic areas surrounding small lytic foci bilaterally in the distal femur and proximal tibia.
A second surgery was done in March 2022 and the bilateral pre-zygomatic masses were removed. Histopathological examination showed features similar to that seen in the previous excision but with low cellularity and increased fibrosis. The histological examination showed groups, clusters and sheets of foamy macrophages set in a dense fibrous tissue with interspersed spindle cells and lymphoid aggregates with scattered giant cells with eosinophilic cytoplasm surrounded by multiple nuclei and clearing of cytoplasm at the periphery (Touton-type giant cells). These are also called xanthelasmatic giant cells due to their association with xanthelasmas. The histiocytic cells were positive for CD68 and negative for CD1a and S100 immunostains. The overall microscopic features were similar to a fibrohistiocytic lesion with an exuberant proliferation of foamy macrophages [Figure 2].
Patient consent was obtained for publication purposes.
Discussion
ECD is a very rare chronic multisystem histiocytic neoplasm. The diagnosis is established by clinical, radiological and histological findings. Histiocytic disorders can be subdivided into Langerhans cell histiocytosis, non-Langerhans histiocytosis and malignant histiocytic disorders.8 Non-Langerhans histiocytoses are derived from the monocyte-macrophage lineage which are positive for CD68 and negative for CD1a. S100 staining is variable. ECD is a non-Langerhans histiocytic disorder characterised by multifocal osteosclerotic lesions of the long bones in addition to organ infiltration.9,10
There are few reported ECD related neoplasms in the retro-orbital area or facial bones, however, this case presented with bilateral facial soft tissue lesions. The reported endocrine abnormalities include hypopituitarism, hypogonadism and hypothyroidism. Hypogonadism is mainly confirmed by fertility issues, and this was seen later in this case who presented with hypogonadism, hypothyroidism and diabetes insipidus.11,12
Skin and soft tissue involvement is mainly in the form of xanthomas and xanthelasmas mostly in the head and neck area, predominantly the periorbital area, which is reported in approximately one-third of patients in multiple series. This was also seen in this case who had bilateral periorbital xanthelasmas.12
As this disease shares some common stem cells with haematopoietic neoplasms, approximately 10% of these tumours are associated with myeloid malignancies, for example, myeloid leukaemia. This must be taken into consideration in this case.13
The main reason for organ damage is fibrosis which results from fibroblastic proliferation in response to lymphokines and cytokines, which induce organ fibrosis and are not caused by the infiltration of histiocytes into the organs.14
Secondary involvement by autoimmune disease is common in ECD; approximately 40% of the cases have SLE-involvement and can have the typical serology for SLE. This is also typical in this case who presented later with features and serology of SLE.15
Treatment of ECD has drastically evolved since the better understanding of molecular aspects of the disease. Targeted therapies such as BRAF inhibitors and MEK inhibitors (drugs that block the MAPK pathway) have promising results yet considerable risks and side effects.16,17
Conclusion
ECD remains a diagnosis of exclusion and should be considered when dealing with patients having multiple bone and soft tissue lesions with suggestive histology and multisystem involvement. Molecular studies hold a key position to make the diagnosis and provide hope for targeted therapy.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chester WÜber Lipoidgranulomatose Virchows Arch Path Anat 1930279561602 10.1007/BF 01942684 · doi ↗
- 2Jaffe HL Gaucher’s disease and certain other inborn metabolic disorders: lipid (cholesterol) granulomatosis Jaffe HL Metabolic and inflammatory disease of the bone and joints Philadelphia, USA Lea & Febiger 1972 535 41
- 3Starkebaum G Hendrie P Erdheim–Chester disease Best Pract Res Clin Rheumatol 2020 34 101510 10.1016/j.berh.2020.101510 32305313 · doi ↗ · pubmed ↗
- 4Emile JF Abla O Fraitag S Horne A Haroche J Donadieu J Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages Blood 2016127267281 10.1182/blood-2016-01-69063626966089 PMC 5161007 · doi ↗ · pubmed ↗
- 5Diamond Eli L Dagna L Hyman DM Cavalli G Janku F Estrada-Veras J Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease Blood 201412448392 10.1182/blood-2014-03-56138124850756 PMC 4110656 · doi ↗ · pubmed ↗
- 6Veyssier-Belot C Cacoub P Caparros-Lefebvre D Wechsler J Brun B Remy M Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases Medicine (Baltimore)19967515769 10.1097/00005792-199605000-000058965684 · doi ↗ · pubmed ↗
- 7Emile JF Diamond ELHélias-Rodzewicz Z Cohen-Aubart F Charlotte F Hyman DM Recurrent RAS and PIK 3CA mutations in Erdheim-Chester disease Blood 2014124301619 10.1182/blood-2014-04-57093725150293 PMC 4224196 · doi ↗ · pubmed ↗
- 8Brousse N Pileri S Haroche J World Health Organization classification of tumours of haematopoietic and lymphoid tissues Swerdlow S Campo E Harris N Jaffe E Pileri S Stein H World Health Organization classification of tumours International Agency for Research on Cancer Lyon, France 2017 481 3