Red Cell Pyruvate Kinase Deficiency With Hypertriglyceridemia: A Case Report
Dinesh V Hinge, Mamta Muranjan, Amar Taksande, Priyanka Hampe

TL;DR
A four-month-old baby with red cell pyruvate kinase deficiency and high triglycerides was successfully treated with early diagnosis and care.
Contribution
Highlights a rare case linking pyruvate kinase deficiency with hypertriglyceridemia and successful management strategies.
Findings
A four-month-old male with PK deficiency and severe hypertriglyceridemia showed improvement with specific treatment.
Early diagnosis and treatment improved prognosis and quality of life in the affected infant.
Lipemia retinitis and splenohepatomegaly were observed as clinical features in this case.
Abstract
Red cell pyruvate kinase (PK) deficiency is a genetic disorder affecting the enzyme PK in red blood cells. A deficiency in PK leads to hemolytic anemia. Hypertriglyceridemia means elevated levels of triglycerides in the blood. The hypertriglyceridemia disorder can be primary or secondary to an underlying disease. Hypertriglyceridemia with β-thalassemia major is a known association and is called hypertriglyceridemia-thalassemia syndrome. A four-month-old male child was found to have milky serum. On investigation, there was severe anemia, with triglycerides at 1197 mg/dL and high lactate dehydrogenase (LDH). The child had severe pallor, mild icterus, a dysmorphic face, and splenohepatomegaly. Ophthalmic examination showed lipemia retinitis. The child was treated with medium-chain fatty acid formula feed. Regular blood transfusions, folic acid supplements, and avoidance of salicylate group…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
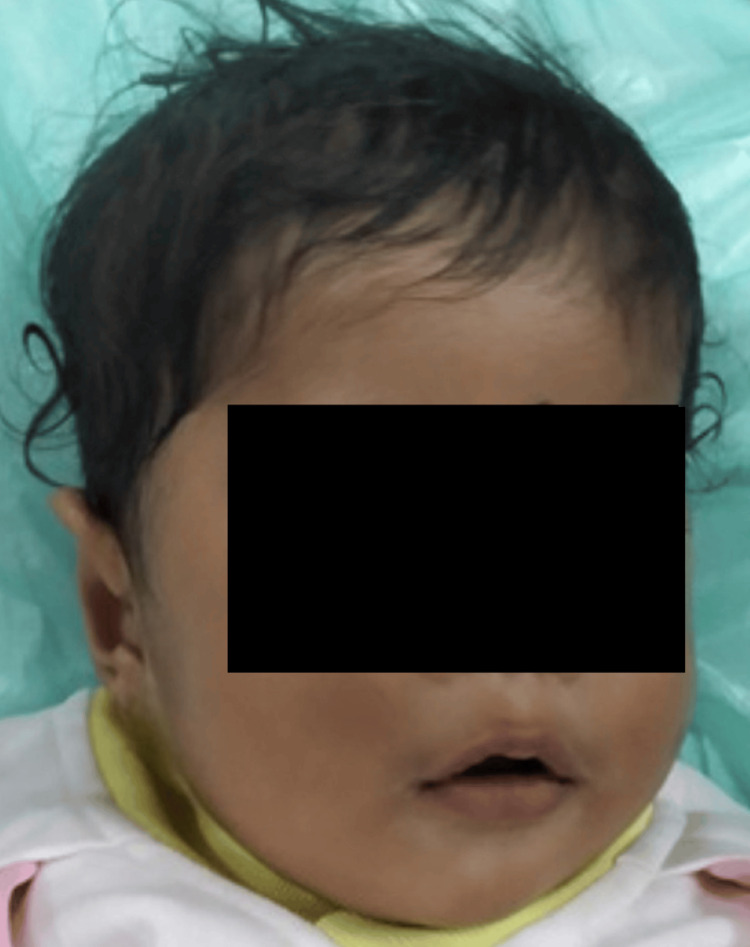 Figure 1
Figure 1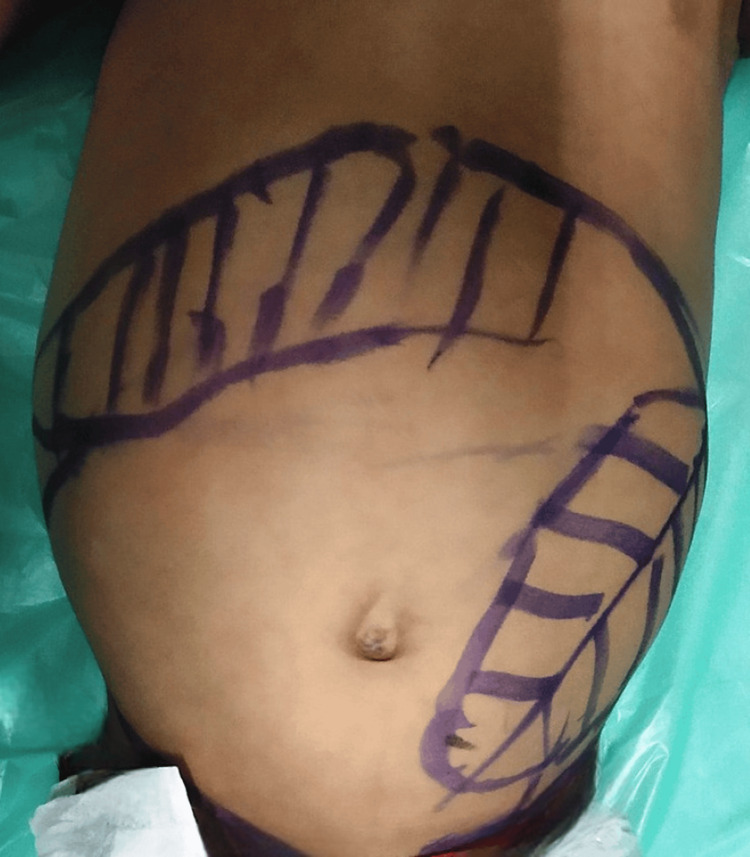 Figure 2
Figure 2 Figure 3
Figure 3| Parameters | On admission at 4months of life | Follow-up at 6 months | Follow-up at 8 months | Follow-up at 12 months |
| Hemoglobin (g/dL) | 6.1 | 4.5 | 4.5 | 8 |
| WBC (cells/μL) | 13000 | 10000 | 11100 | 9500 |
| Platelet (cells/μL) | 2.6 | 1.2 | 1.8 | 2.2 |
| Bilirubin (total/direct) (mg/dL) | 2.2/0.36 | 2/0.7 | 1.84/0.57 | 2.2/0.6 |
| SGOT/SGPT (U/L) | 29/32 | 58/18 | 37/18 | 45/32 |
| Triglyserides (mg/dL) | 1197 | 907 | 281 | 280 |
| Cholesterol (mg/dL) | 117 | 100 | 117 | 124 |
| Serum LDH (U/L) | 3091.2 | 2400 | 700 | 432 |
| Human red blood cell enzymes | Results | Normal range |
| Red cell pyruvate kinase | 4.7 IU/g Hb | 8-14 IU/g Hb |
| Glucose-6-phosphate dehydrogenase | 6.27 IU/g Hb | 4-13 IU/g Hb |
| Glucose phosphate isomerase | 64.6 IU/g Hb | 45-75 IU/g Hb |
| Red cell using eosin-5′-maleimide | 925.0 MCF | 900-1300 MCF |
| Fractions | Results (mg/dL) | Reference range (%) |
| Alpha-lipoproteins | 8 | =<46.0 |
| Pre-B-lipoproteins | 41.9 | =<29.0 |
| Beta-lipoproteins | 50.1 | 41-72 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsErythrocyte Function and Pathophysiology · Neonatal Health and Biochemistry · Hemoglobin structure and function
Introduction
Red cell pyruvate kinase (PK) deficiency is the most predominant glycolytic pathway abnormality that causes hereditary non-spherocytic hemolytic anemia. The clinical manifestation varies, ranging from minor symptoms to critically ill newborn anemia and jaundice that need to be treated in intensive care [1]. It is an autosomal recessive condition caused by a PK-LR gene mutation [1]. The presentation will be on severe neonatal anemia, chronic hemolysis, and neonatal jaundice [2]. Treatment of this disease is primarily supportive: blood transfusion, splenectomy, and definitive treatment like bone marrow transplant [2]. Acute or chronic hemolysis is associated with hypertriglyceridemia, which can be acquired or hereditary and exhibits enhanced synthesis or reduced catabolism [3]. Hypertriglyceridemia can be primary or secondary. Primary familial hypertriglyceridemia is autosomal dominant. Secondary hemolysis causes transient hyperlipidemia through direct red cell destruction or intravascular coagulation [4]. The association between hypertriglyceridemia and red cell PK deficiency is seen in the medical literature.
Case presentation
A four-month-old male child, born of third-degree consanguinity, presented with pallor since birth and abdominal distension for the past two months. The baby was born by normal vaginal delivery. After 12 hours of life, the baby developed severe respiratory distress and indirect hyperbilirubinemia. Respiratory support was provided, and an exchange transfusion was performed for neonatal hyperbilirubinemia, followed by two days of phototherapy. The child has a history of two hospitalizations for low hemoglobin, during which packed red blood cell transfusions were administered. On head-to-toe examination, the child exhibited severe pallor and mild icterus. Additionally, dysmorphic facial features and frontal bossing were observed, as shown in Figure 1.
Dysmorphic face and frontal bossing
Weight and height were on the third centile with normal head circumference. Systemic examination revealed spleenohepatomegaly (spleen size 10 cm, liver size 6 cm), as shown in Figure 2.
Enlarged spleen and liver size (splenohepatomegaly)
The cardiovascular and central nervous system were normal. On fundus examination, there were signs suggestive of lipemia retinalis. A hemogram showed a white blood cell count of 13000 cells/μL, Hb of 6.1 g/dL, Hct of 18.8%, reticulocyte count of 6.7%, and platelet count of 260000 cells/μL, as shown in Table 1.
The peripheral blood smear revealed polychromasia and nucleated erythrocytes. Direct and indirect Coombs’ tests were negative; the red blood cell membrane study was normal. The red blood cell enzyme study showed PK deficiency, with red cell PK activity at 4.7 IU/g, as shown in Table 2.
Serum lactate dehydrogenase (LDH) was 3091 U/L, suggesting hemolysis. Renal and liver function were normal, and high-performance liquid chromatography (HPLC) and bone marrow examination were normal. During blood sampling, we noticed a milky blood sample (Figure 3).
Milky blood sample
Hence, a lipid profile was obtained: serum triglyceride was 1197 mg/dL, and cholesterol was 119 mg/dL. Serum lipoprotein electrophoresis is suggestive of type IV hypertriglyceridemia, as shown in Table 3.
Ultrasonography showed splenohepatomegaly with normal liver texture. Ophthalmological examination revealed lipemia retinitis. Appropriate testing was used to rule out other causes of secondary hypertriglyceridemia, such as hypothyroidism, diabetes mellitus, and nephrotic syndrome. The mother had no laboratory evidence of hyperlipidemia. The father had died in a road traffic accident. A mutation study was advised to confirm the PK-LR gene mutation, but it was not done due to financial constraints. The child was given medium-chain fatty acid formula feed along with breastfeeding, blood transfusion, and folic acid supplements, and was advised to avoid salicylate groups of drugs. The lipid profile and complete blood count were repeated after two months, showing decreased serum triglycerides (564 mg/dL) and serum cholesterol (124 mg/dL) with low hemoglobin, so a blood transfusion was given. On examination, regression of spleen and liver size was noted. The child is receiving regular blood transfusions. Consequently, weight gain has been noted, and the child is developmentally normal.
Discussion
The most common enzymopathy is red cell PK deficit, followed by glucose-6-phosphate dehydrogenase deficiency. It is caused by a mutation in a single gene, PK-LR [1]. Mutations in the PK-LR gene influence both the liver and red blood cell isozymes; however, only red cell glycolysis is severely compromised since mature erythrocytes are unable to replace damaged PK through fresh synthesis; only hepatocytes can. Hereditary disorders cause primary hypertriglyceridemia. Secondary hypertriglyceridemia is caused by high-fat diets, diabetes, obesity, hypothyroidism, and certain drugs (tamoxifen and estrogen). It is considered that epigenetic, environmental, and genetic factors contribute to the wide range of clinical manifestations. In this case report of red cell PK deficiency with severe hypertriglyceridemia, secondary hypertriglyceridemia is uncommon in individuals with chronic hemolytic anemia [3]. The known cases have been children with thalassemia major, which is associated with hypertriglyceridemia-thalassemia syndrome [5].
In other cases, serum triglyceride levels returned to normal after red cell transfusion therapy [5]. Malnutrition is also linked to hypertriglyceridemia in young children with thalassemia or PK deficiency [3]. Zieve syndrome (hyperlipidemia, jaundice, and transitory hemolytic anemia caused by alcohol abuse) demonstrates acquired PK deficiencies [6]. In a study of 36 patients with hemolysis, 27 had hypertriglyceridemia with hemolytic crisis; causes of hemolysis included microangiopathic hemolytic anemia, autoimmune hemolysis, infection, and glucose-6-phosphate dehydrogenase deficiency [4]. The case report of an eight-month-old female with thalassemia major and hypertriglyceridemia demonstrated clinical characteristics like classical thalassemia, except for a milky plasma [7]. This case shows the rare association of red cell PK deficiency with hypertriglyceridemia; it is usually resolved by dietary modification and blood transfusion therapy. The underlying pathogenesis is challenging to understand and requires molecular study.
Conclusions
Early diagnosis and dietary modification can improve the prognosis and maintain a near-normal life for affected infants. Regular blood transfusions every month are required to maintain normal hemoglobin levels in PK deficiency disorder. Our case represents an unusual association of red cell PK deficiency with hypertriglyceridemia. This association could be coincidental or part of the same disease process. Molecular testing will be essential for a definitive diagnosis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Red cell pyruvate kinase deficiency: molecular and clinical aspects Br J Haematol Zanella A Fermo E Bianchi P Valentini G 112513020051598234010.1111/j.1365-2141.2005.05527.x · doi ↗ · pubmed ↗
- 2Successful bone marrow transplantation in a child with red blood cell pyruvate kinase deficiency Bone Marrow Transplant Tanphaichitr VS Suvatte V Issaragrisil S 6896902620001104157210.1038/sj.bmt.1702576 · doi ↗ · pubmed ↗
- 3Severe hypertriglyceridemia in an infant with red cell pyruvate kinase deficiency Indian Pediatr Rao A Hulbert M Wilson DB 303305442007 https://pubmed.ncbi.nlm.nih.gov/17468529/17468529 · pubmed ↗
- 4Hyperlipidemia in acute hemolysis Klin Wochenschr Druml W Grimm G Laggner AN Schneeweiss B Lenz K 426429691991194295410.1007/BF 01666827 · doi ↗ · pubmed ↗
- 5Hypertriglyceridemia thalassemia syndrome: common disease, uncommon association Indian J Pediatr Das L Samprathi M Shukla U Bandyopadhyay D Das RR 7207228320162693520110.1007/s 12098-016-2062-x · doi ↗ · pubmed ↗
- 6Red cell metabolic and membrane features in haemolytic anaemia of alcoholic liver disease (Zieve's syndrome)Br J Haematol Goebel KM Goebel FD Schubotz R Schneider J 57358535197787140910.1111/j.1365-2141.1977.tb 00622.x · doi ↗ · pubmed ↗
- 7Hypertriglyceridemia thalassemia syndrome J Pediatr Endocrinol Metab Jain M Ali W Singh BB Verma N Kumar A 8218223120182990215610.1515/jpem-2018-0171 · doi ↗ · pubmed ↗